- Published on
Emergency and Acute Medicine – Osteoporosis
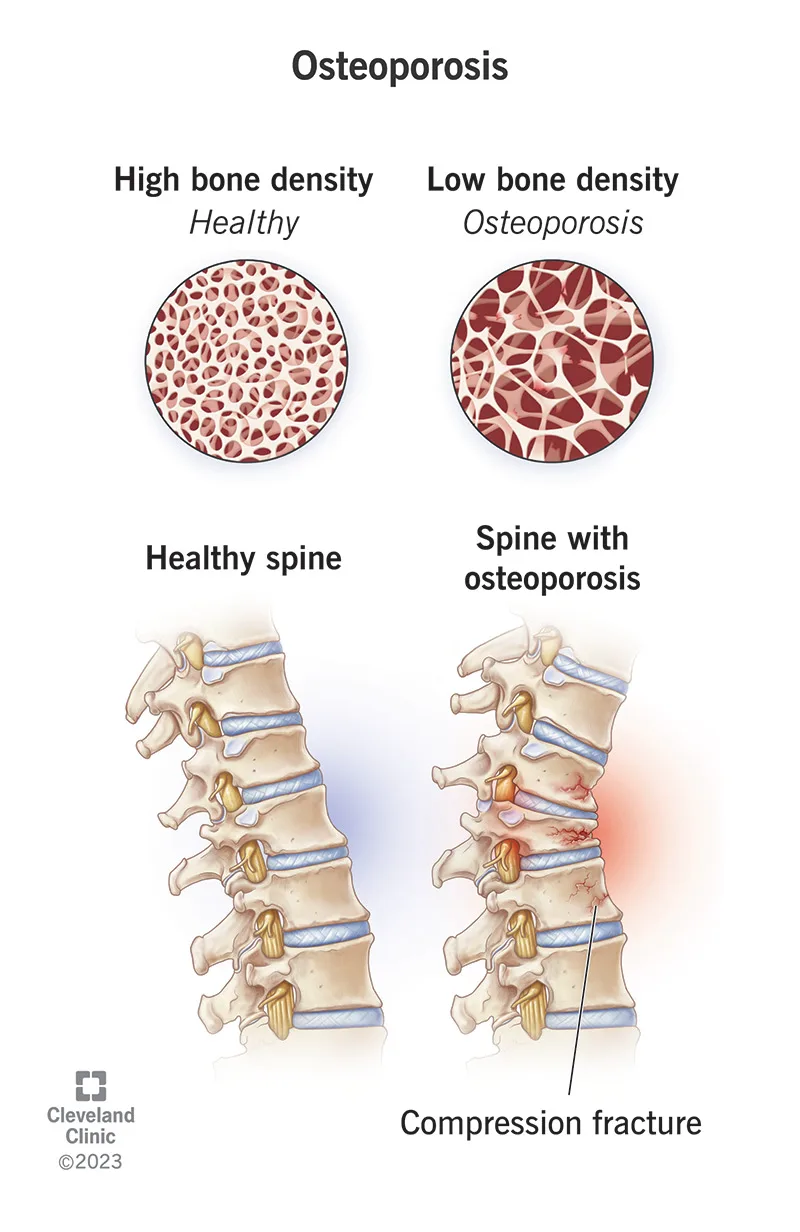
Osteoporosis is characterized by a diffuse decrease in skeletal mass, with trabecular bone—particularly in the vertebrae and femur—affected earlier and more severely than cortical bone. Although the disease process begins in adolescence, fractures typically do not manifest until age 50 years or older. Females are affected far more commonly than males, especially following menopause.
The underlying mechanism is an imbalance between bone resorption and bone formation, with resorption predominating. Advanced age is the most significant risk factor. Additional contributors include inadequate dietary calcium intake, particularly early in life, a sedentary lifestyle with lack of weight-bearing activity, and estrogen deficiency after menopause. Other important risk factors include long-term corticosteroid use, alcoholism, methotrexate therapy, tobacco use, low body weight, and familial predisposition. Pediatric patients are generally asymptomatic despite early onset of bone loss.
Osteoporosis is usually clinically silent until pathologic fractures occur. Fractures resulting from minimal trauma or recurrent fractures are hallmark features. The vertebral column is most commonly involved, with multiple compression fractures often leading to kyphosis and scoliosis. Hip fractures, including femoral neck and intertrochanteric fractures, are also frequent. A history of fractures after low-energy mechanisms or a family history of osteoporosis should raise suspicion. Physical examination findings typically relate to the acute fracture rather than the underlying disease itself.
Evaluation focuses on identifying fractures without significant mechanisms and assessing risk factors. A careful neurovascular examination distal to femoral or other extremity fractures is essential. In patients with vertebral fractures, rectal tone and postvoid residual should be assessed. Plain radiographs of suspected fractures may show osteopenia, though this is a late finding. Spine films may reveal old compression fractures. Computed tomography is recommended for vertebral fractures to assess for retropulsion and spinal canal compromise, which may not be evident on plain films.
Laboratory studies such as serum calcium, parathyroid hormone, and alkaline phosphatase can help differentiate osteoporosis from other metabolic bone disorders. Imaging with plain radiographs identifies fractures, but determining fracture age can be difficult. Bone scan or CT may help clarify fracture chronicity, particularly in the spine. Bone densitometry using dual-energy x-ray absorptiometry confirms the diagnosis, with a bone mineral density T-score of −2.5 or lower defining osteoporosis.
The differential diagnosis includes multiple myeloma or metastatic malignancy, osteogenesis imperfecta, hyperparathyroidism, and other demineralizing bone diseases. Management in the emergency setting prioritizes fracture care. Prehospital providers should minimize patient movement and report mechanism details suggestive of pathologic fracture. Initial treatment involves immobilization and standard fracture management, recognizing that healing may be delayed or incomplete. Orthotic back braces may be needed for vertebral fractures in consultation with orthopedics. Prevention remains more effective than acute treatment, and long-term therapy should be initiated or reinforced.
Pharmacologic therapy includes bisphosphonates such as alendronate or risedronate as first-line agents, with alternatives including zoledronic acid, raloxifene, denosumab, calcitonin, parathyroid hormone analogs, and, in selected cases, estrogen therapy. Adequate calcium and vitamin D supplementation are essential. Admission decisions follow standard orthopedic protocols, with particular attention to age, mobility, neurologic symptoms, and social factors. Compression fractures are often stable, but cervical fractures or those with neurologic deficits require admission and emergent specialty consultation.
Key points include recognizing recurrent or low-energy fractures as suggestive of osteoporosis, understanding that osteopenia on plain radiographs is a late but important clue, and initiating referral for definitive evaluation and treatment. Bisphosphonates remain first-line therapy for long-term management, and coordinated follow-up is critical to prevent future fractures.
- Published on
KembaraXtra-Emergency and Acute Medicine - Osteomyelitis
Description: Osteomyelitis is an infection of bone characterized by ongoing inflammatory destruction. It is most commonly bacterial in origin, although fungal osteomyelitis can occur, particularly in immunocompromised patients. The disease may present as acute, subacute, or chronic infection, with chronic cases defined by persistence or recurrence and the presence of necrotic bone (sequestrum).
Etiology: Hematogenous osteomyelitis occurs when bacteria seed bone via the bloodstream and is most common in children, the elderly, and people who inject drugs. Children typically develop acute disease, often without a preceding illness, though up to one-third report recent trauma. Adults more often develop subacute or chronic disease. Staphylococcus aureus is the most common pathogen across all age groups. Neonates are additionally affected by Enterobacteriaceae, group A and B streptococci, and Escherichia coli. Children may also develop infection from Haemophilus influenzae, while Salmonella is classically associated with sickle cell disease. Adults may have infections caused by gram-negative rods, Pseudomonas, Staphylococcus epidermidis, and anaerobes. Vertebral osteomyelitis is uncommon but typically affects adults over 45 years, often in the setting of diabetes, malignancy, hemodialysis, long-term catheterization, or IV drug use, and may extend to cause epidural abscesses or deep paraspinal collections. Direct or contiguous osteomyelitis occurs following trauma, open fractures, surgery, or spread from adjacent soft tissue infection and is more common in adults and adolescents. Chronic osteomyelitis is associated with necrotic bone and commonly involves S. aureus, S. epidermidis, Pseudomonas aeruginosa, and gram-negative organisms.
Clinical features: Symptoms vary with disease duration. Patients often present with localized, deep, dull, or throbbing bone pain that may occur at rest or with movement. Fever and chills may be present in acute disease but are often absent in chronic infection. Other features include malaise, nausea, vomiting, reluctance to use an affected limb, nonhealing ulcers, or fracture nonunion. Risk factors include diabetes mellitus, vascular disease, IV drug use, trauma, and invasive procedures. Examination may reveal localized warmth, erythema, edema, tenderness, decreased range of motion, sinus tract drainage, or exposed bone. Deep ulcers with palpable bone and a positive “probe-to-bone” test strongly suggest osteomyelitis.
Evaluation: Initial workup includes complete blood count, erythrocyte sedimentation rate, C-reactive protein, plain radiographs, and blood and wound cultures. Leukocytosis may be absent, but inflammatory markers are usually elevated. Blood cultures are positive in approximately half of cases. Plain radiographs are often normal in the first two to three weeks; early findings include periosteal elevation, followed by cortical erosion and new bone formation. MRI is the imaging modality of choice, with high sensitivity and specificity, allowing early detection and assessment of marrow, cortical, and soft tissue involvement. CT is useful when MRI is contraindicated and for surgical planning. Bone scans and leukocyte scintigraphy may be helpful in selected cases, while ultrasound is increasingly useful in children. Definitive diagnosis is established by bone biopsy with histology and culture, which remains the gold standard.
Management: Initial management focuses on stabilization, particularly in septic patients or those with neurologic deficits from spinal involvement. Empiric intravenous antibiotics should be started after cultures are obtained, then tailored based on organism and sensitivities. Treatment typically requires four to six weeks of parenteral antibiotics, with shorter IV courses followed by oral therapy possible in selected pediatric cases. Orthopedic and infectious disease consultation is essential, and surgical intervention is often required for debridement of necrotic bone, infected hardware, or abscesses.
Disposition and follow-up: Patients with acute osteomyelitis should be admitted for intravenous antibiotics and monitoring. Chronic osteomyelitis often requires admission for surgical management and prolonged therapy. Selected subacute or chronic cases may be managed as outpatients if debridement has been performed, cultures obtained, and reliable home IV antibiotic therapy is available. Close follow-up is mandatory to monitor response and prevent recurrence.
Key points: A normal white blood cell count does not exclude osteomyelitis. Early radiographs may be normal, making MRI critical for early diagnosis. Wound cultures alone are often unreliable for guiding therapy, and bone biopsy provides the most accurate microbiologic diagnosis.


- Published on
KembaraXtra-Emergency and Acute Medicine - Osteogenesis Imperfecta
Description: Osteogenesis imperfecta is an inherited disorder caused by abnormalities in the procollagen amino acid sequence, leading to defective collagen formation. This results in bone hypomineralization, incomplete ossification, and brittle bones. Because collagen is a major component of connective tissue, multiple organ systems may be affected. The clinical course is variable, with most patients experiencing recurrent fractures during childhood followed by relative quiescence during adolescence and early adulthood.
Etiology: The condition arises from defects in procollagen that disrupt the structure of bone and connective tissue matrix. Mutations at different sites on the procollagen protein chain produce varying disease severity. Inheritance patterns include autosomal recessive forms, which are generally milder, and autosomal dominant forms, which are typically more severe. Lethal variants often result from sporadic or new mutations. Related disorders such as Ehlers–Danlos syndrome involve mutations in the same collagen protein but at different locations.
Clinical features: The hallmark manifestation is recurrent fractures, often occurring with minimal or trivial trauma, particularly in long bones. Fractures may be present at birth, recur throughout childhood, or reappear in the elderly. Skeletal abnormalities include bowed or shortened limbs, pectus excavatum, scoliosis, kyphosis, vertebral compression fractures, and abnormal skull shape. Blue sclerae are a classic finding and are not associated with visual impairment. Hearing loss, usually sensorineural, commonly begins in adolescence, with most patients affected by early adulthood. Dental abnormalities such as discolored, fragile, or misshapen teeth are frequent. Joint laxity, valvular disease, vascular abnormalities, and occasional thyroid dysfunction may also occur. Severe cases may result in perinatal death.
Pediatric considerations: In children, repeated fractures or fractures from low-impact mechanisms should raise suspicion for osteogenesis imperfecta. However, nonaccidental trauma must always be considered, and a careful social history and thorough evaluation are essential to distinguish between the two.
Evaluation: Diagnosis is typically based on a combination of clinical findings and radiographic features. A history of multiple fractures or fractures inconsistent with the reported mechanism is suggestive. A careful physical examination should assess for tenderness at other sites, blue sclerae, dental abnormalities, joint laxity, and neurovascular status distal to fractures.
Investigations: Laboratory studies are used to exclude metabolic causes of bone fragility, including abnormalities in calcium, phosphate, vitamin D, vitamin C, or parathyroid hormone levels. Genetic testing may be performed for confirmation, family counseling, or prenatal diagnosis. Radiographs often demonstrate osteopenia, crumpled or bowed long bones, incomplete ossification at physes, and characteristic findings such as wormian bones of the skull. A skeletal survey is mandatory in children. Audiologic testing should be arranged in older patients.
Management: There is no definitive cure for osteogenesis imperfecta. Acute management in the emergency setting focuses on stabilization, pain control, and appropriate immobilization of fractures. Fracture management is determined by injury type and location, with early orthopedic consultation for consideration of traction or operative fixation when indicated.
Disposition and follow-up: Admission is based on injury severity, presence of multiple fractures, or need for operative intervention. Pediatric patients may require admission to evaluate for possible nonaccidental trauma. Patients with isolated fractures and adequate home support may be managed as outpatients with close orthopedic and primary care follow-up. Long-term management focuses on monitoring disease progression, preventing fractures, and addressing hearing and mobility issues.
Key points: Osteogenesis imperfecta should be suspected in patients, especially children, with recurrent fractures or fractures from minor trauma. Differentiating pathologic fractures from nonaccidental trauma is critical and often challenging. Pain perception is normal in these patients, and respiratory infections may occur more frequently due to chest wall abnormalities.


- Published on
KembaraXtra-Emergency and Acute Medicine - Osgood–Schlatter Disease
Description: Osgood–Schlatter disease is the most common cause of knee pain in children aged 10–15 years and represents a benign, self-limited extra-articular condition. It is characterized by pain, swelling, and tenderness over the tibial tuberosity at the insertion of the patellar tendon, just below the knee joint. Symptoms are activity related, worsening with exercise and improving with rest, and are commonly seen in physically active adolescents during periods of rapid growth.
Etiology: The most widely accepted mechanism involves repetitive traction and microfractures at the tibial tubercle apophysis caused by repeated stress from the patellar tendon. Activities that involve running, jumping, and sudden changes in direction increase strain on the extensor mechanism and precipitate symptoms.
Clinical features: Patients present with localized pain and swelling over the tibial tuberosity that is exacerbated by running, jumping, kneeling, or climbing stairs and relieved by rest. The condition is usually unilateral, although bilateral involvement occurs in approximately 20% of cases. Risk factors include age between 10 and 15 years, male sex, pubertal growth spurts, and participation in sports such as soccer, basketball, volleyball, and skating.
Physical examination: Examination reveals prominence, tenderness, and soft tissue swelling over the tibial tuberosity with pain reproduced by resisted knee extension. Quadriceps and hamstring tightness is common compared with the unaffected side. Mild erythema may be present, but the knee joint examination itself is otherwise normal, with no effusion or instability.
Evaluation: Diagnosis is primarily clinical based on history and examination. Imaging is not routinely required but may be obtained if the diagnosis is uncertain or to exclude other pathology. Plain knee radiographs may show fragmentation or irregular ossification of the tibial tuberosity, while ultrasound can demonstrate associated soft tissue changes.
Management: Treatment is conservative and focuses on symptom control and activity modification. Patients should rest from painful activities for approximately 6–8 weeks, particularly avoiding jumping and cutting sports. Ice application, stretching of the quadriceps and hamstrings, and use of analgesics such as ibuprofen or acetaminophen are recommended. An infrapatellar tendon strap or protective padding may reduce strain during activities. Corticosteroid injections should be avoided, and reassurance is essential as the condition resolves with skeletal maturity.
Disposition and follow-up: Admission is not required, and patients can be safely discharged home. Follow-up with a pediatrician or primary care provider in 2–3 weeks is advised to reassess symptoms and activity tolerance. Referral to pediatric orthopedics is rarely necessary and is reserved for patients

- Published on
KembaraXtra-Emergency and Acute Medicine - Organophosphate Poisoning
Description: Organophosphate poisoning results from irreversible inhibition of cholinesterase enzymes, particularly acetylcholinesterase, leading to accumulation of acetylcholine at central and peripheral synapses and subsequent cholinergic overdrive. Toxic effects involve muscarinic, nicotinic, and central nervous system pathways, which may overlap in presentation. Mortality is primarily due to respiratory failure caused by bronchorrhea, bronchoconstriction, respiratory muscle weakness, and central respiratory depression. In children, symptoms may be difficult to distinguish, and seizures occur more frequently than in adults.
Etiology: Exposure commonly occurs through agricultural insecticides or chemical nerve agents such as sarin, soman, tabun, and VX. These agents are rapidly absorbed through the lungs, gastrointestinal tract, skin, mucous membranes, and eyes, making even dermal exposure potentially life-threatening.
Clinical features: The classic presentation is the cholinergic toxidrome characterized by DUMBELS: diarrhea and diaphoresis, urination, miosis and muscle fasciculations, bradycardia with bronchorrhea and bronchospasm, emesis, lacrimation, and salivation. Mild exposure causes headache, dizziness, weakness, tremors, and anorexia. Moderate exposure leads to muscle fasciculations progressing to flaccid paralysis, respiratory muscle weakness, agitation, confusion, pinpoint pupils, nausea, vomiting, and excessive secretions. Severe exposure presents with seizures, coma, centrally mediated respiratory depression, bronchoconstriction, cyanosis, cardiac conduction abnormalities, profuse secretions, and urinary or fecal incontinence.
Evaluation: A focused history should assess occupational exposure, recent insecticide use, possible suicide attempt, and access to pesticides, with retrieval of the original container if available. Physical examination emphasizes identification of parasympathetic signs, muscle weakness, and respiratory compromise. Diagnosis is clinical, and treatment should not be delayed for laboratory confirmation.
Diagnostic testing: Red blood cell cholinesterase levels best reflect synaptic inhibition but are often delayed, while plasma cholinesterase levels are more rapidly available but less specific. Severity correlates with the degree of enzyme inhibition, though therapy must begin immediately regardless of results. Additional testing includes CBC, electrolytes, renal function, glucose, arterial blood gas when respiratory symptoms are present, ECG for dysrhythmias or heart block, and chest radiography if pulmonary edema or aspiration is suspected.
Management: Initial management prioritizes decontamination and protection of healthcare workers using appropriate personal protective equipment. All contaminated clothing should be removed and double-bagged, and skin thoroughly washed with soap and water. Airway protection, oxygenation, and ventilatory support are critical, with early intubation when indicated. Atropine is the primary antidote for muscarinic symptoms and should be administered in escalating doses every five minutes until bronchial secretions are dry, without using pupil size or heart rate as treatment endpoints. Pralidoxime should be given early to reverse nicotinic effects and regenerate cholinesterase before enzyme aging occurs, improving muscle strength and reducing paralysis. Supportive care includes frequent suctioning, avoidance of succinylcholine during intubation, and cautious gastric decontamination in early severe ingestions.
Disposition and follow-up: Any symptomatic patient or those requiring atropine should be admitted, typically to an intensive care setting, for close monitoring. Asymptomatic patients may be discharged after 6–12 hours of observation with clear return precautions and reliable follow-up. Intentional exposures require psychiatric evaluation, and poison control or toxicology consultation is recommended for significant or ongoing symptoms.
Key points: Inadequate atropine dosing is the most common cause of treatment failure. Clinical diagnosis should guide therapy without delay for laboratory confirmation. Recognition and aggressive management of respiratory compromise are essential to reduce mortality.

- Published on
KembaraXtra-Emergency and Acute Medicine - Optic Neuritis
Description: Optic neuritis is an inflammatory disorder of the optic nerve resulting in demyelination and acute optic nerve dysfunction. It is strongly associated with multiple sclerosis and is the presenting manifestation in approximately 15–20% of patients with MS. Inflammation may involve the optic disc (papillitis) or the retrobulbar portion of the optic nerve, where the funduscopic examination may initially appear normal. The long-term risk of developing clinically definite MS depends on MRI findings, with significantly higher risk in patients demonstrating multiple demyelinating lesions.
Etiology and risk factors: Most cases are idiopathic and self-limited, but 20–50% are associated with multiple sclerosis. Other causes include postviral inflammation following infections such as varicella, measles, mononucleosis, HSV, or VZV, typically occurring weeks after illness. Granulomatous and infectious causes include tuberculosis, syphilis, sarcoidosis, cryptococcosis, Lyme disease, and HIV-related infections. Drug-induced optic neuritis has been reported with amiodarone, ethambutol, and tamoxifen. Genetic predisposition is suggested by associations with HLA-A23, B7, and DR2 alleles.
Clinical features: Patients typically present with subacute vision loss developing over days, peaking within 1–2 weeks, most often unilateral in adults and bilateral in children. Retrobulbar pain, worsened by eye movement, is characteristic. Color vision, contrast sensitivity, and depth perception are disproportionately affected compared with visual acuity. An afferent pupillary defect is common in unilateral cases. Visual field testing often reveals a central scotoma. Funduscopic examination may show optic disc swelling or appear normal. Uhthoff phenomenon, transient worsening of vision with heat or exertion, may occur.
Evaluation: A detailed history should assess age, sex, onset and progression of visual loss, eye pain, prior neurologic symptoms, recent infections, drug exposure, and family history of MS. Physical examination requires a complete ophthalmologic and neurologic assessment including visual acuity, pupillary reflexes, color vision testing, visual fields, and dilated fundus examination. Blood pressure should be assessed to exclude hypertensive optic neuropathy.
Diagnostic testing: MRI of the brain and orbits with gadolinium is the imaging modality of choice and demonstrates optic nerve enhancement in the majority of acute cases while also stratifying future MS risk. CT is less sensitive and primarily used to exclude compressive lesions. Laboratory evaluation is guided by clinical suspicion and may include CBC, ESR, syphilis serology, Lyme testing, ANA, HIV testing, and tuberculosis screening. Chest radiography may assist in evaluating sarcoidosis or tuberculosis. Formal automated visual field testing is recommended for baseline assessment and follow-up.
Management: Early ophthalmology and neurology consultation is essential. High-dose IV corticosteroids followed by an oral taper are recommended for patients with severe visual loss or those with two or more demyelinating lesions on MRI, as this shortens recovery time and reduces short-term risk of MS progression. Oral corticosteroids alone should be avoided, as they increase recurrence risk. Treatment decisions should be individualized in patients with fewer MRI lesions.
Disposition and follow-up: Admission is indicated for bilateral vision loss, diagnostic uncertainty, or when IV steroid therapy is required. Patients with unilateral involvement, stable condition, and reliable follow-up may be discharged with urgent neurology and ophthalmology review. High-risk patients should be referred for disease-modifying therapy consideration. Prompt follow-up is mandatory, as MRI findings are the strongest predictor of future multiple sclerosis.
Key points: Space-occupying lesions must be excluded before diagnosing optic neuritis. Acute bilateral visual loss with headache or diplopia raises concern for alternative emergencies such as pituitary apoplexy. MRI is critical for prognostication, and management should be coordinated with specialists to align with current standards of care.

- Published on
KembaraXtra-Emergency and Acute Medicine - Opportunistic Infections
Description: Opportunistic infections are unusual infections that occur when host defenses are impaired, allowing normally nonpathogenic organisms to cause disease. They are most commonly associated with advanced immunosuppression and may present with subtle or atypical signs, yet progress rapidly to severe systemic illness.
Etiology: In patients with HIV/AIDS, opportunistic infections typically occur when the CD4 T-lymphocyte count falls below 200 cells/mm³ or <14% of total lymphocytes. common pathogens include pneumocystis jiroveci (pcp), disseminated tuberculosis, toxoplasma gondii, cryptococcus, histoplasma, cytomegalovirus, mycobacterium avium complex, jc virus (progressive multifocal leukoencephalopathy), hepatitis b virus, and human herpesvirus-8 (kaposi sarcoma). cell-mediated immune deficiency from hematologic malignancies, lymphoma, high-dose glucocorticoids, autoimmune disease, chemotherapy, radiation, or viral infections predisposes to such as legionella, nocardia, salmonella, mycobacteria. neutrophil impairment depletion due cytotoxic drugs, aplastic anemia, marrow infiltration, drug reactions, vitamin deficiencies increases risk for with staphylococcus, α-hemolytic streptococcus, enteric organisms, anaerobes, invasive aspergillosis.< />pan>
Clinical features: Patients may present with new or worsening fatigue, fever or hypothermia, chills, night sweats, tachypnea, and signs of systemic inflammatory response. Pulmonary sources cause cough, dyspnea, and rales; genitourinary sources cause dysuria, frequency, or retention; gastrointestinal sources cause abdominal pain, vomiting, diarrhea, bleeding, or jaundice; and CNS involvement may cause confusion, headache, focal neurologic deficits, or seizures. Ambulatory hypoxia is characteristic of PCP pneumonia, and oropharyngeal candidiasis is a key marker of immune suppression.
Evaluation: A thorough history is essential, including known HIV status and CD4 count, malignancy or transplant history, autoimmune disease, and use of cytotoxic or high-dose steroid therapy. Physical examination must be comprehensive, as classic signs of infection may be minimal or absent. Fever or any clinical deterioration should prompt full evaluation.
Diagnostic testing: Laboratory evaluation includes CBC with differential to identify leukocytosis or neutropenia, calculation of absolute neutrophil count, blood and site-specific cultures, urinalysis, electrolytes, renal function, glucose, lactate, coagulation studies, and LDH (often elevated in PCP). If CD4 count is unknown, an absolute lymphocyte count <1,000 />mu;L predicts CD4 <200. imaging includes chest radiography, which may show nonspecific infiltrates or bilateral interstitial changes in pcp, and high-resolution ct of the for early pcp detection. head with contrast is indicated focal neurologic deficits suspected toxoplasmosis, abdominal pelvic warranted when a gastrointestinal source suspected. lumbar puncture required cns infection suspected, diagnostic paracentesis should be performed immunocompromised patients ascites.< />pan>
Management: Initial stabilization focuses on airway, breathing, and circulation, with supplemental oxygen, IV access, fluid resuscitation for hypotension, cardiac monitoring, and early empiric antimicrobial therapy. Broad-spectrum antibiotics are initiated promptly, often using antipseudomonal penicillins with aminoglycosides or monotherapy with third- or fourth-generation cephalosporins, fluoroquinolones, or carbapenems when indicated. Vancomycin is added in areas with high prevalence of resistant organisms. Antifungal therapy is initiated if there is no improvement after adequate antibacterial coverage, and trimethoprim–sulfamethoxazole is first-line therapy for suspected PCP, with alternatives for intolerance. Corticosteroids are indicated in PCP with hypoxemia and should be started within 72 hours of antimicrobial therapy.
Disposition and follow-up: All patients with suspected or confirmed systemic opportunistic infections require hospital admission. Discharge is appropriate only when systemic infection has been confidently excluded. Infectious disease consultation is strongly recommended. Clinicians must maintain a high index of suspicion, as immunocompromised patients may present with minimal signs yet deteriorate rapidly.

- Published on
KembaraXtra -Medicine- Neonatal Sepsis
Neonatal sepsis is a life-threatening systemic infection occurring in newborns, most commonly within the first month of life and rarely up to three months of age. It is predominantly bacterial in origin, with pathogens typically acquired from maternal perineal flora during or around delivery. The incidence is approximately 3–5 cases per 1,000 live births, and despite advances in care, neonatal sepsis remains a major cause of morbidity and mortality. Early recognition is critical because clinical signs are often subtle and rapidly progressive.
The etiology is most commonly bacterial, with Group B Streptococcus, Escherichia coli, and Listeria monocytogenes being the leading pathogens. Other bacterial causes include coagulase-positive and coagulase-negative Staphylococcus species and Treponema pallidum. Viral causes, though less common, include herpes simplex virus, enteroviruses, adenovirus, and cytomegalovirus, while fungal infections such as Candida species are typically seen in premature or immunocompromised infants. Infection may progress to meningitis, which carries a higher risk of long-term neurologic sequelae.
Risk factors are divided into perinatal and neonatal categories. Perinatal risk factors include maternal fever, urinary tract infection, chorioamnionitis, prolonged rupture of membranes exceeding 18 hours, foul lochia, uterine tenderness, and intrapartum asphyxia. Neonatal risk factors include prematurity, fetal tachycardia, male sex, twinning, congenital immune or metabolic disorders such as galactosemia, congenital anomalies, omphalitis, and prior intramuscular iron administration. The presence of any risk factor should significantly lower the threshold for evaluation.
Clinical presentation is often nonspecific. Caregivers may report poor feeding, lethargy, or irritability. On examination, infants may appear toxic with temperature instability, apnea, bradycardia, tachycardia, tachypnea, prolonged capillary refill, mottled or cyanotic skin, abdominal distention, jaundice, or bleeding. Severe disease may progress to septic shock, hypoglycemia, seizures, disseminated intravascular coagulation, cardiovascular collapse, and death if untreated.
Evaluation requires a full sepsis workup followed immediately by empiric antibiotic therapy. Bedside glucose measurement is essential. Laboratory studies include a complete blood count, inflammatory markers such as C-reactive protein, blood cultures, catheterized or suprapubic urine cultures, and cerebrospinal fluid studies when the infant is stable. Additional tests include arterial blood gas analysis, electrolytes, calcium levels, coagulation studies when indicated, and chest radiography to assess for pneumonia.
Management begins with stabilization of airway, breathing, and circulation, including ventilatory support when needed and prompt intravenous access. Empiric antibiotics are initiated immediately, most commonly ampicillin combined with gentamicin or cefotaxime, with vancomycin added if clinical deterioration or resistant organisms are suspected. Supportive care for shock, metabolic derangements, and organ dysfunction is provided as required.
All infants with suspected neonatal sepsis must be admitted for intravenous antibiotics, close monitoring, and supportive care. In practice, any infant younger than one month presenting with fever is generally admitted even if suspicion for sepsis is low. Prognosis depends on the timeliness of treatment, the causative organism, and the presence of complications, emphasizing the importance of early recognition and aggressive management.
- Published on
KembaraXtra -Medicine- Nephritic Syndrome
Nephritic syndrome represents acute glomerulonephritis, an inflammatory injury of the glomeruli characterized by abrupt onset of hematuria, often cola- or coffee-colored, with or without red blood cell casts, variable proteinuria, reduced urine output, edema, hypertension, azotemia, and possible acute renal failure. The underlying mechanism is incompletely understood but involves immune-mediated injury to the glomerular basement membrane with crescent formation reflecting severe capillary wall damage.
Poststreptococcal glomerulonephritis is a classic cause and follows infection with group A β-hemolytic streptococci, occurring as a nonsuppurative complication after pharyngitis or skin infection. A latent period of 1–3 weeks after pharyngeal infection or 2–4 weeks after cutaneous infection helps distinguish it from IgA nephropathy. Immune complex deposition produces characteristic subepithelial “hump-shaped” deposits, with low complement C3 levels lasting 6–8 weeks. Prognosis is excellent in most patients, particularly children, with spontaneous recovery of renal function within weeks, although transient nephrotic features may occur during recovery.
Other infectious causes include sepsis, pneumonia, endocarditis, viral infections including HIV, and parasitic diseases such as malaria. Hepatitis virus–associated glomerular disease may present with nephritic or nephrotic features and typically shows persistently low complement levels. Noninfectious immune-mediated causes include systemic lupus erythematosus, Henoch–Schönlein purpura, vasculitis, Goodpasture syndrome, and IgA nephropathy, which is the most common cause worldwide and typically presents with hematuria coinciding with upper respiratory infections and normal complement levels.
Patients commonly present with hematuria, periorbital or generalized edema, hypertension, and symptoms related to recent infection. Children may develop facial edema, whereas older adults may present with congestive heart failure. Nonspecific symptoms such as malaise, anorexia, nausea, and weakness are common. Physical examination often reveals elevated blood pressure and fluid overload.
Evaluation centers on urinalysis showing hematuria, proteinuria, and red blood cell casts, which are diagnostic of active glomerular inflammation. Laboratory studies include complete blood count, renal function tests, serum albumin, complement levels, streptococcal antibody titers, and cultures when infection is suspected. Renal ultrasound assesses kidney size, and chest radiography may reveal pulmonary edema. Renal biopsy is not routinely required for poststreptococcal disease but is indicated for atypical features, persistent hypocomplementemia, severe proteinuria, or progressive renal dysfunction.
Management focuses on treating the underlying cause and controlling complications. Streptococcal infections are treated with appropriate antibiotics. Salt and fluid restriction, loop diuretics for edema, and antihypertensive therapy with ACE inhibitors or ARBs are used to control blood pressure and proteinuria. Severe cases with uremia, electrolyte disturbances, refractory fluid overload, or acidosis require hemodialysis. Hypertensive emergencies are managed with titratable intravenous antihypertensives.
Hospital admission is required for patients with oliguria, rising creatinine, significant edema, electrolyte abnormalities, severe hypertension, heart failure, or suspected systemic disease. Mild cases in otherwise healthy patients may be managed with close outpatient follow-up and nephrology referral. Prognosis is generally favorable, but long-term monitoring of blood pressure, renal function, and proteinuria is essential, particularly in IgA nephropathy and rapidly progressive forms.
- Published on
KembaraXtra -Medicine- Nephrotic Syndrome
Nephrotic syndrome is a clinical disorder caused by defects in the glomerular filtration barrier that lead to heavy protein loss in the urine. It is defined by nephrotic-range proteinuria greater than 3 g per 24 hours, hypoalbuminemia with serum albumin below 3 g/dL, peripheral edema, hyperlipidemia, lipiduria with oval fat bodies and fatty casts, and associated hypogammaglobulinemia. The altered glomerular basement membrane becomes excessively permeable due to immune complexes, nephrotoxic antibodies, or nonimmune mechanisms, allowing loss of albumin and other large proteins.
The pathophysiology centers on increased glomerular protein filtration and reduced plasma oncotic pressure, resulting in sodium and water retention and edema. Severe hypoalbuminemia may cause postural hypotension, syncope, or shock. Hyperlipidemia develops as hepatic lipoprotein synthesis increases in response to low oncotic pressure. Patients are at increased risk of thromboembolic events, particularly when serum albumin is very low, protein excretion is high, fibrinogen levels are elevated, and antithrombin III levels are reduced.
Nephrotic syndrome may arise from primary renal diseases or secondary systemic conditions. In adults, membranous nephropathy is the most common primary cause and may be associated with chronic infections such as hepatitis B or C or autoimmune disease. Minimal change disease accounts for most pediatric cases and typically responds well to corticosteroids. Focal segmental glomerulosclerosis affects adolescents and young adults and may be primary or secondary to conditions such as HIV, obesity, or heroin use. Diabetic nephropathy is the most common secondary cause in adults and is heralded by microalbuminuria. Other causes include systemic lupus erythematosus, monoclonal gammopathies such as amyloidosis and multiple myeloma, HIV-associated nephropathy, preeclampsia, hepatitis, and drug-induced disease.
Clinical presentation varies widely. Many patients are asymptomatic aside from foamy urine and gradual weight gain from fluid retention. Edema may range from mild pitting edema to generalized anasarca with ascites and pleural effusions. Hypertension or hypotension may occur depending on intravascular volume status. Hematuria may be present, particularly with renal vein thrombosis. Acute dyspnea, tachycardia, or hypotension should raise concern for pulmonary embolism, a recognized complication of the hypercoagulable state.
Evaluation begins with urinalysis demonstrating heavy proteinuria and lipiduria with oval fat bodies. Laboratory studies reveal hypoalbuminemia, hyperlipidemia, and possible anemia. Quantification of proteinuria is performed with a 24-hour urine collection or protein-to-creatinine ratio. Additional testing is guided by suspected systemic disease and may include autoimmune markers, hepatitis serologies, and serum or urine protein electrophoresis. Renal ultrasound is useful in secondary causes, and renal biopsy is the definitive diagnostic test when response to steroids is poor or etiology is unclear.
Management focuses on treating the underlying cause and controlling complications. Sodium restriction and cautious diuresis with loop diuretics are used to manage edema, avoiding overly aggressive fluid removal to prevent renal failure and thrombosis. ACE inhibitors or ARBs reduce proteinuria and slow progression of renal disease. Corticosteroids are the mainstay for primary nephrotic syndromes, particularly minimal change disease. Anticoagulation is indicated for confirmed thromboembolic events and may be considered prophylactically in patients with profound hypoalbuminemia. Lipid-lowering therapy and nutritional support are important adjuncts.
Hospital admission is required for patients with severe edema, respiratory compromise, acute renal failure, thromboembolic complications, or significant comorbid disease. Stable patients without complications may be managed as outpatients with close nephrology follow-up. Long-term prognosis depends on the underlying cause, degree of proteinuria, blood pressure control, and response to therapy, with vigilant monitoring essential to reduce complications and progression to chronic kidney disease.

