- Published on
Emergency and Acute Medicine – Otitis Media
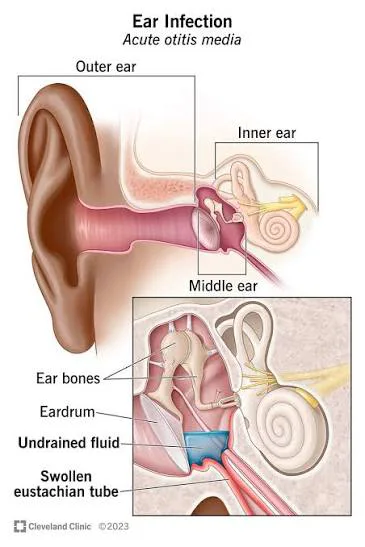
Otitis media is an inflammatory condition of the middle ear and is one of the most common infections encountered in early childhood, particularly between 6 and 36 months of age. It is characterized by a rapid onset of local and systemic symptoms and has a high recurrence rate, with more than one-third of children experiencing more than five episodes by the age of seven. Although it predominantly affects children, otitis media can also occur in adults.
The condition most often develops in association with upper respiratory tract infections, which lead to eustachian tube dysfunction and impaired drainage of the middle ear. Viral pathogens commonly implicated include parainfluenza virus, respiratory syncytial virus, influenza virus, adenovirus, and rhinovirus. Bacterial causes include Streptococcus pneumoniae, Moraxella catarrhalis, Haemophilus influenzae, Streptococcus pyogenes, and Mycoplasma pneumoniae. Predisposing factors include impaired mucociliary function, immunodeficiency, nasotracheal intubation, and certain populations such as American Indians, Eskimos, and individuals with Down syndrome, cleft palate, diabetes, vitamin A deficiency, or HIV. Additional risk factors include daycare attendance, family history, exposure to parental smoking, pacifier use, and bottle-feeding.
Diagnosis is primarily clinical and follows the American Academy of Pediatrics 2013 guidelines. Otitis media should be diagnosed when there is moderate to severe bulging of the tympanic membrane, mild bulging of the tympanic membrane accompanied by recent onset of ear pain, or new otorrhea not attributable to otitis externa. The diagnosis should not be made in the absence of middle ear effusion, which is best assessed using pneumatic otoscopy or tympanometry. Recurrent otitis media is defined as three episodes within six months or four episodes within one year, with at least one episode occurring in the preceding six months.
Patients typically present with ear pain, irritability, fever, and symptoms of an upper respiratory infection such as rhinorrhea. Younger children may exhibit poor feeding, vomiting, diarrhea, or tugging at the ear, while older children and adults may complain of a plugged-ear sensation, vertigo, tinnitus, or conjunctivitis. On physical examination, the tympanic membrane often appears erythematous, inflamed, and bulging, with decreased mobility and obscured landmarks. New-onset otorrhea in the absence of otitis externa is also a key finding.
The essential evaluation includes careful otoscopic examination with full visualization of the tympanic membrane and assessment of its mobility. Laboratory testing is generally unnecessary, and cultures are not helpful unless obtained via tympanocentesis. Imaging, such as computed tomography, is reserved for suspected complications like mastoiditis. Tympanocentesis may be indicated in cases of severe pain or toxicity, failure of antimicrobial therapy, suspected suppurative complications, illness in neonates, or immunocompromised patients.
Management depends on age, severity, and laterality of infection. Many mild cases resolve spontaneously without antibiotics. Antibiotic therapy is recommended for all infants younger than six months, children younger than two years with bilateral otitis media, children older than six months with severe symptoms (persistent otalgia for more than 48 hours or fever ≥102.2°F), and cases with tympanic membrane rupture and drainage. In otherwise healthy children aged six months or older with mild symptoms or uncertain diagnosis, a period of observation with close follow-up in two to three days is appropriate, provided caregivers are reliable. Antipyretics and analgesics are essential for symptom control, while antihistamines, decongestants, and steroids have not shown proven benefit.
First-line antibiotic therapy typically includes high-dose amoxicillin, with amoxicillin–clavulanate or alternative agents such as azithromycin or cefuroxime used based on allergy history, recent antibiotic use, or local resistance patterns. Parenteral antibiotics are reserved for toxic-appearing infants, immunocompromised patients, or those unable to tolerate oral therapy.
Most patients can be safely discharged with appropriate treatment and follow-up. Admission is indicated for febrile or toxic children younger than one year, immunocompromised patients, those with significant dehydration, inability to tolerate oral intake, suspected serious associated infection, unreliable caregivers, or concern for abuse. Follow-up is recommended within 10–14 days to ensure resolution, with earlier reassessment if symptoms fail to improve within 24–48 hours or worsen.
Important complications of otitis media include recurrent infections, tympanic membrane perforation, serous otitis media, hearing loss, facial nerve injury, mastoiditis, cholesteatoma, meningitis, and other intracranial infections. A key clinical pearl is that in otherwise healthy children aged six months or older with mild symptoms, observation without immediate antibiotics and close follow-up is often a safe and effective approach.
Otitis media is an inflammatory condition of the middle ear and is one of the most common infections encountered in early childhood, particularly between 6 and 36 months of age. It is characterized by a rapid onset of local and systemic symptoms and has a high recurrence rate, with more than one-third of children experiencing more than five episodes by the age of seven. Although it predominantly affects children, otitis media can also occur in adults.
The condition most often develops in association with upper respiratory tract infections, which lead to eustachian tube dysfunction and impaired drainage of the middle ear. Viral pathogens commonly implicated include parainfluenza virus, respiratory syncytial virus, influenza virus, adenovirus, and rhinovirus. Bacterial causes include Streptococcus pneumoniae, Moraxella catarrhalis, Haemophilus influenzae, Streptococcus pyogenes, and Mycoplasma pneumoniae. Predisposing factors include impaired mucociliary function, immunodeficiency, nasotracheal intubation, and certain populations such as American Indians, Eskimos, and individuals with Down syndrome, cleft palate, diabetes, vitamin A deficiency, or HIV. Additional risk factors include daycare attendance, family history, exposure to parental smoking, pacifier use, and bottle-feeding.
Diagnosis is primarily clinical and follows the American Academy of Pediatrics 2013 guidelines. Otitis media should be diagnosed when there is moderate to severe bulging of the tympanic membrane, mild bulging of the tympanic membrane accompanied by recent onset of ear pain, or new otorrhea not attributable to otitis externa. The diagnosis should not be made in the absence of middle ear effusion, which is best assessed using pneumatic otoscopy or tympanometry. Recurrent otitis media is defined as three episodes within six months or four episodes within one year, with at least one episode occurring in the preceding six months.
Patients typically present with ear pain, irritability, fever, and symptoms of an upper respiratory infection such as rhinorrhea. Younger children may exhibit poor feeding, vomiting, diarrhea, or tugging at the ear, while older children and adults may complain of a plugged-ear sensation, vertigo, tinnitus, or conjunctivitis. On physical examination, the tympanic membrane often appears erythematous, inflamed, and bulging, with decreased mobility and obscured landmarks. New-onset otorrhea in the absence of otitis externa is also a key finding.
The essential evaluation includes careful otoscopic examination with full visualization of the tympanic membrane and assessment of its mobility. Laboratory testing is generally unnecessary, and cultures are not helpful unless obtained via tympanocentesis. Imaging, such as computed tomography, is reserved for suspected complications like mastoiditis. Tympanocentesis may be indicated in cases of severe pain or toxicity, failure of antimicrobial therapy, suspected suppurative complications, illness in neonates, or immunocompromised patients.
Management depends on age, severity, and laterality of infection. Many mild cases resolve spontaneously without antibiotics. Antibiotic therapy is recommended for all infants younger than six months, children younger than two years with bilateral otitis media, children older than six months with severe symptoms (persistent otalgia for more than 48 hours or fever ≥102.2°F), and cases with tympanic membrane rupture and drainage. In otherwise healthy children aged six months or older with mild symptoms or uncertain diagnosis, a period of observation with close follow-up in two to three days is appropriate, provided caregivers are reliable. Antipyretics and analgesics are essential for symptom control, while antihistamines, decongestants, and steroids have not shown proven benefit.
First-line antibiotic therapy typically includes high-dose amoxicillin, with amoxicillin–clavulanate or alternative agents such as azithromycin or cefuroxime used based on allergy history, recent antibiotic use, or local resistance patterns. Parenteral antibiotics are reserved for toxic-appearing infants, immunocompromised patients, or those unable to tolerate oral therapy.
Most patients can be safely discharged with appropriate treatment and follow-up. Admission is indicated for febrile or toxic children younger than one year, immunocompromised patients, those with significant dehydration, inability to tolerate oral intake, suspected serious associated infection, unreliable caregivers, or concern for abuse. Follow-up is recommended within 10–14 days to ensure resolution, with earlier reassessment if symptoms fail to improve within 24–48 hours or worsen.
Important complications of otitis media include recurrent infections, tympanic membrane perforation, serous otitis media, hearing loss, facial nerve injury, mastoiditis, cholesteatoma, meningitis, and other intracranial infections. A key clinical pearl is that in otherwise healthy children aged six months or older with mild symptoms, observation without immediate antibiotics and close follow-up is often a safe and effective approach.
- Published on
KembaraXtra- Medicine – Atrial Flutter
Typical atrial flutter is a stable macroreentrant atrial rhythm that circulates around the tricuspid annulus in the right atrium. The critical component of this circuit is the cavotricuspid isthmus (CTI), the tissue between the inferior vena cava and the tricuspid valve, which is why typical flutter is also referred to as CTI-dependent atrial flutter. Because the circuit is anatomically and physiologically stable, atrial depolarization is regular, usually occurring at a rate of 250–350 beats per minute.
Atypical atrial flutter refers to regular macroreentrant atrial tachyarrhythmias that do not depend on the CTI. These forms often occur after cardiac surgery, in patients with congenital heart disease, or following catheter ablation procedures—particularly left atrial ablation for atrial fibrillation—although they may also occur without an identifiable cause.
Conduction through the atrioventricular (AV) node in atrial flutter often follows predictable ratios. For example, with an atrial rate of 300 beats per minute, 2:1 AV conduction produces a ventricular rate of about 150 beats per minute, while 3:1 and 4:1 conduction produce ventricular rates of approximately 100 and 75 beats per minute, respectively. When AV nodal conduction varies, the ventricular rhythm may appear irregular, although a consistent atrial cycle length is often still present.
Atrial flutter is the second most common sustained atrial tachyarrhythmia after atrial fibrillation, with an estimated 200,000 new cases each year in the United States. Its prevalence increases with age and is about 2.5 times more common in men than in women. It is frequently seen in patients with congestive heart failure, chronic obstructive pulmonary disease, pulmonary embolism, pulmonary hypertension, or in the early postoperative period following open-heart surgery. Importantly, more than half of patients with atrial flutter will develop atrial fibrillation within three years, and more than 80% within five years.
Clinically, atrial flutter may present with palpitations, dizziness, light-headedness, syncope or near syncope, chest pain, dyspnea, or worsening heart failure. Some patients develop thromboembolic complications due to intracardiac thrombus formation. Common etiologic associations include age-related atrial degeneration, rheumatic or congenital heart disease, ventricular dysfunction, mitral valve disease, thyrotoxicosis, pulmonary embolism, obesity, pericarditis, and prior cardiac surgery. Antiarrhythmic therapy for atrial fibrillation may also predispose patients to developing atrial flutter.
Diagnosis is primarily established by electrocardiography. Typical findings include absence of normal P waves and the presence of regular “sawtooth” flutter waves without an isoelectric baseline, most clearly seen in the inferior leads (II, III, and aVF). Atrioventricular conduction is usually 2:1, 3:1, or 4:1 rather than 1:1, unless pre-excitation is present. Holter or event monitoring may be useful for intermittent symptoms or to assess rate control. Echocardiography is recommended in newly diagnosed patients to evaluate for structural heart disease, and transesophageal echocardiography may be required to exclude atrial thrombus before cardioversion. Electrophysiologic studies are used for definitive diagnosis and for catheter ablation therapy.
Typical atrial flutter is a stable macroreentrant atrial rhythm that circulates around the tricuspid annulus in the right atrium. The critical component of this circuit is the cavotricuspid isthmus (CTI), the tissue between the inferior vena cava and the tricuspid valve, which is why typical flutter is also referred to as CTI-dependent atrial flutter. Because the circuit is anatomically and physiologically stable, atrial depolarization is regular, usually occurring at a rate of 250–350 beats per minute.
Atypical atrial flutter refers to regular macroreentrant atrial tachyarrhythmias that do not depend on the CTI. These forms often occur after cardiac surgery, in patients with congenital heart disease, or following catheter ablation procedures—particularly left atrial ablation for atrial fibrillation—although they may also occur without an identifiable cause.
Conduction through the atrioventricular (AV) node in atrial flutter often follows predictable ratios. For example, with an atrial rate of 300 beats per minute, 2:1 AV conduction produces a ventricular rate of about 150 beats per minute, while 3:1 and 4:1 conduction produce ventricular rates of approximately 100 and 75 beats per minute, respectively. When AV nodal conduction varies, the ventricular rhythm may appear irregular, although a consistent atrial cycle length is often still present.
Atrial flutter is the second most common sustained atrial tachyarrhythmia after atrial fibrillation, with an estimated 200,000 new cases each year in the United States. Its prevalence increases with age and is about 2.5 times more common in men than in women. It is frequently seen in patients with congestive heart failure, chronic obstructive pulmonary disease, pulmonary embolism, pulmonary hypertension, or in the early postoperative period following open-heart surgery. Importantly, more than half of patients with atrial flutter will develop atrial fibrillation within three years, and more than 80% within five years.
Clinically, atrial flutter may present with palpitations, dizziness, light-headedness, syncope or near syncope, chest pain, dyspnea, or worsening heart failure. Some patients develop thromboembolic complications due to intracardiac thrombus formation. Common etiologic associations include age-related atrial degeneration, rheumatic or congenital heart disease, ventricular dysfunction, mitral valve disease, thyrotoxicosis, pulmonary embolism, obesity, pericarditis, and prior cardiac surgery. Antiarrhythmic therapy for atrial fibrillation may also predispose patients to developing atrial flutter.
Diagnosis is primarily established by electrocardiography. Typical findings include absence of normal P waves and the presence of regular “sawtooth” flutter waves without an isoelectric baseline, most clearly seen in the inferior leads (II, III, and aVF). Atrioventricular conduction is usually 2:1, 3:1, or 4:1 rather than 1:1, unless pre-excitation is present. Holter or event monitoring may be useful for intermittent symptoms or to assess rate control. Echocardiography is recommended in newly diagnosed patients to evaluate for structural heart disease, and transesophageal echocardiography may be required to exclude atrial thrombus before cardioversion. Electrophysiologic studies are used for definitive diagnosis and for catheter ablation therapy.

- Published on
KembaraXtra-Medicine – Atrial Myxoma
Atrial myxoma is a benign neoplasm of mesenchymal origin and represents the most common primary tumor of the heart. Although histologically benign, atrial myxomas are clinically significant because their size, mobility, and intracardiac location can lead to serious and sometimes life-threatening complications. They are most commonly referred to as cardiac myxomas.
Primary cardiac tumors are extremely rare, with an autopsy prevalence ranging from 0.001% to 0.3%, and metastatic tumors occur far more frequently than primary tumors. Among primary cardiac tumors, myxomas account for 30% to 50% of benign cases. Approximately 65% occur in females. Most cases are sporadic, but 4.5% to 10% are familial, commonly associated with Carney complex. The median age of presentation is about 56 years, although familial cases tend to occur earlier, with an average age of 25 years. Most myxomas arise in the left atrium (about 75%), followed by the right atrium, right ventricle, and left ventricle. They are typically pedunculated and attached to the interatrial septum.
Histopathologically, atrial myxomas are characterized by abundant loose myxoid stroma containing scattered round, polygonal, or stellate cells with dense, irregular nuclei derived from multipotent mesenchymal cells. While benign in classification, myxomas can secrete cytokines and growth factors, contributing to systemic and constitutional symptoms in addition to mechanical complications.
Clinically, patients with atrial myxomas usually present in one of three ways: atrioventricular valve obstruction, systemic embolization, or constitutional symptoms. Obstructive symptoms may mimic mitral or tricuspid valve disease and include dyspnea, orthopnea, paroxysmal nocturnal dyspnea, syncope, dizziness, edema, atrial fibrillation, or rarely sudden death. Symptoms that vary with body position, particularly improvement when recumbent, are suggestive. Systemic embolization occurs in nearly one third of patients and may manifest as stroke, pulmonary embolism, paradoxical embolism, or acute coronary syndrome. Constitutional features such as fever, weight loss, arthralgia, and Raynaud phenomenon are also common, while rare presentations include peripheral neuropathy, vasculitis, or paraneoplastic syndromes. On examination, findings may include murmurs, signs of pulmonary hypertension, a widely split first heart sound, or a characteristic early diastolic “tumor plop.”
Most atrial myxomas are sporadic, but familial cases are commonly associated with Carney complex, an autosomal dominant condition characterized by cardiac and extracardiac myxomas, pigmented skin lesions, endocrine hyperactivity, and other tumors such as schwannomas. Multiple genetic loci and mutations have been identified in this syndrome.
Diagnosis requires a high index of suspicion due to nonspecific symptoms that overlap with many cardiovascular and pulmonary conditions. Laboratory tests may show anemia, thrombocytopenia, elevated inflammatory markers, increased immunoglobulins, or elevated cardiac biomarkers, though these findings are nonspecific. Electrocardiography may reveal atrial enlargement, arrhythmias, or conduction abnormalities. Transthoracic echocardiography is the initial diagnostic test of choice, with approximately 95% sensitivity, while transesophageal echocardiography approaches 100% sensitivity and better defines tumor attachment and mobility. CT and MRI are valuable for delineating tumor size, extension, vascularity, and for distinguishing myxoma from atrial thrombus. Cardiac catheterization may demonstrate neovascularization and is sometimes used to assess for concomitant coronary artery disease before surgery.
The definitive treatment for atrial myxoma is prompt surgical excision, which should not be delayed due to the risk of embolization or sudden death. Postoperatively, arrhythmias or conduction disturbances may occur and are managed accordingly. In rare cases, particularly with recurrent tumors associated with Carney complex, advanced surgical approaches such as cardiac autotransplantation or transplantation may be required.
Prognosis after surgical excision is excellent, with reported survival rates of approximately 95% at three years. However, recurrence can occur in up to 5% of sporadic cases and up to 20% of familial cases, particularly within the first six years. Risk factors for recurrence include familial disease, atypical tumor location, and multicentric tumors. Malignant transformation into atrial myxofibrosarcoma, although rare, should be considered in cases of early recurrence. Untreated atrial myxomas carry a significant risk of sudden death, estimated at up to 15%.
Referral to a cardiologist is recommended for diagnosis and management, and once identified, early consultation with a cardiovascular surgeon is essential. Long-term follow-up with periodic echocardiography is advised to monitor for recurrence, especially in patients with familial disease or Carney complex.
Atrial myxoma is a benign neoplasm of mesenchymal origin and represents the most common primary tumor of the heart. Although histologically benign, atrial myxomas are clinically significant because their size, mobility, and intracardiac location can lead to serious and sometimes life-threatening complications. They are most commonly referred to as cardiac myxomas.
Primary cardiac tumors are extremely rare, with an autopsy prevalence ranging from 0.001% to 0.3%, and metastatic tumors occur far more frequently than primary tumors. Among primary cardiac tumors, myxomas account for 30% to 50% of benign cases. Approximately 65% occur in females. Most cases are sporadic, but 4.5% to 10% are familial, commonly associated with Carney complex. The median age of presentation is about 56 years, although familial cases tend to occur earlier, with an average age of 25 years. Most myxomas arise in the left atrium (about 75%), followed by the right atrium, right ventricle, and left ventricle. They are typically pedunculated and attached to the interatrial septum.
Histopathologically, atrial myxomas are characterized by abundant loose myxoid stroma containing scattered round, polygonal, or stellate cells with dense, irregular nuclei derived from multipotent mesenchymal cells. While benign in classification, myxomas can secrete cytokines and growth factors, contributing to systemic and constitutional symptoms in addition to mechanical complications.
Clinically, patients with atrial myxomas usually present in one of three ways: atrioventricular valve obstruction, systemic embolization, or constitutional symptoms. Obstructive symptoms may mimic mitral or tricuspid valve disease and include dyspnea, orthopnea, paroxysmal nocturnal dyspnea, syncope, dizziness, edema, atrial fibrillation, or rarely sudden death. Symptoms that vary with body position, particularly improvement when recumbent, are suggestive. Systemic embolization occurs in nearly one third of patients and may manifest as stroke, pulmonary embolism, paradoxical embolism, or acute coronary syndrome. Constitutional features such as fever, weight loss, arthralgia, and Raynaud phenomenon are also common, while rare presentations include peripheral neuropathy, vasculitis, or paraneoplastic syndromes. On examination, findings may include murmurs, signs of pulmonary hypertension, a widely split first heart sound, or a characteristic early diastolic “tumor plop.”
Most atrial myxomas are sporadic, but familial cases are commonly associated with Carney complex, an autosomal dominant condition characterized by cardiac and extracardiac myxomas, pigmented skin lesions, endocrine hyperactivity, and other tumors such as schwannomas. Multiple genetic loci and mutations have been identified in this syndrome.
Diagnosis requires a high index of suspicion due to nonspecific symptoms that overlap with many cardiovascular and pulmonary conditions. Laboratory tests may show anemia, thrombocytopenia, elevated inflammatory markers, increased immunoglobulins, or elevated cardiac biomarkers, though these findings are nonspecific. Electrocardiography may reveal atrial enlargement, arrhythmias, or conduction abnormalities. Transthoracic echocardiography is the initial diagnostic test of choice, with approximately 95% sensitivity, while transesophageal echocardiography approaches 100% sensitivity and better defines tumor attachment and mobility. CT and MRI are valuable for delineating tumor size, extension, vascularity, and for distinguishing myxoma from atrial thrombus. Cardiac catheterization may demonstrate neovascularization and is sometimes used to assess for concomitant coronary artery disease before surgery.
The definitive treatment for atrial myxoma is prompt surgical excision, which should not be delayed due to the risk of embolization or sudden death. Postoperatively, arrhythmias or conduction disturbances may occur and are managed accordingly. In rare cases, particularly with recurrent tumors associated with Carney complex, advanced surgical approaches such as cardiac autotransplantation or transplantation may be required.
Prognosis after surgical excision is excellent, with reported survival rates of approximately 95% at three years. However, recurrence can occur in up to 5% of sporadic cases and up to 20% of familial cases, particularly within the first six years. Risk factors for recurrence include familial disease, atypical tumor location, and multicentric tumors. Malignant transformation into atrial myxofibrosarcoma, although rare, should be considered in cases of early recurrence. Untreated atrial myxomas carry a significant risk of sudden death, estimated at up to 15%.
Referral to a cardiologist is recommended for diagnosis and management, and once identified, early consultation with a cardiovascular surgeon is essential. Long-term follow-up with periodic echocardiography is advised to monitor for recurrence, especially in patients with familial disease or Carney complex.

- Published on
Emergency and Acute Medicine – Lymphadenitis
Basics description
Lymphadenitis refers to inflammation and enlargement of lymph nodes, most commonly as part of a systemic response to infection. Nodes become engorged with lymphocytes and macrophages and may be secondarily involved from infection in a distal extremity, producing painful, tender adenopathy proximally. Acute suppurative lymphadenitis may follow pharyngeal or skin infections and can progress to abscess formation.
Etiology
Lymphadenitis is most frequently caused by bacterial infection. The most common organisms in pyogenic lymphadenitis are Staphylococcus aureus, including community-associated methicillin-resistant S. aureus (CA-MRSA), and group A β-hemolytic Streptococcus. CA-MRSA risk factors include prior MRSA infection, household exposure, military service, incarceration, contact sports, injection drug use, and men who have sex with men. Cervical lymphadenitis usually originates from pharyngeal or periodontal infections and commonly involves streptococci and anaerobes. Axillary lymphadenitis is often caused by group A streptococcus. Nosocomial MRSA should be suspected in patients with recent hospitalization, surgery, dialysis, vascular catheters, recent antibiotic use, or unresponsive infection. In children, acute unilateral cervical suppurative lymphadenitis is most common in those younger than six years and is typically caused by S. aureus, group A streptococcus, or anaerobes.
Diagnosis signs and symptoms
Patients typically present with painful swelling and inflammation of affected lymph nodes, often in association with cellulitis or abscess if the cause is pyogenic. Axillary lymphadenitis may present with fever, axillary pain, and acute lymphedema of the arm or chest and may be associated with ipsilateral pleural effusion. History should include duration of lymphadenopathy, pain, fever, night sweats, weight loss, fatigue, sore throat, cough, occupational and animal exposures, sexual history, drug use, and travel. Physical examination should assess whether lymphadenopathy is localized or generalized, node size, tenderness, overlying skin changes, presence of skin lesions, splenomegaly, and involvement of supraclavicular or scalene nodes, which is always abnormal.
Essential workup
Acute regional lymphadenitis is usually a clinical diagnosis and often part of a broader infectious syndrome such as cellulitis. History and physical examination should focus on identifying an infectious source.
Diagnosis tests and interpretation
Laboratory testing is not always required. A CBC may show leukocytosis with left shift or be normal. Serologic testing for EBV, CMV, HIV, or other pathogens should be guided by clinical suspicion. Ultrasound or CT imaging is indicated when patients fail to improve with therapy or when suppuration is suspected. Percutaneous needle aspiration or surgical drainage should be considered if abscess formation occurs or if there is poor clinical response.
Differential diagnosis
The differential diagnosis includes common infections such as adenovirus, scarlet fever, cat scratch disease, fungal infections, and herpes zoster, as well as unusual infections including sporotrichosis, diphtheria, plague, anthrax, typhoid, rubella, and West Nile virus. Sexually transmitted infections, systemic infections such as HIV, infectious mononucleosis, toxoplasmosis, tuberculosis, hepatitis, and dengue should be considered. Noninfectious causes include drug reactions, malignancy, rheumatologic disorders, and pediatric-specific conditions such as Kawasaki disease and PFAPA syndrome.
Treatment
Initial management includes ensuring airway, breathing, and circulation stability. Treatment is directed at the underlying cause and should account for local resistance patterns, including CA-MRSA prevalence. Outpatient therapy typically lasts 7–10 days and includes limb elevation, moist heat, analgesics, and antibiotics. Abscesses require drainage with culture when possible. Skin-source infections are commonly treated with oral cephalexin plus trimethoprim–sulfamethoxazole or alternatives such as clindamycin or doxycycline. Pharyngeal or periodontal sources are treated with penicillin VK or alternatives such as clindamycin or amoxicillin–clavulanate. Inpatient therapy may require IV penicillin-based regimens with MRSA coverage using vancomycin or clindamycin when indicated.
Disposition and follow-up
Admission is indicated for toxic-appearing patients, those with immunosuppression or significant comorbidities, inability to tolerate oral therapy, or unreliable follow-up. Patients with mild infection who are nontoxic, can take oral antibiotics, and have reliable follow-up within 24–48 hours may be discharged. Failure to resolve promptly with antibiotics should prompt evaluation for malignancy or other serious causes, and lymph node biopsy may be indicated for persistent, large, or supraclavicular nodes.
Pearls and pitfalls
Staphylococcus species are the most common cause of acute regional pyogenic lymphadenitis. Empiric antibiotic therapy should include coverage for CA-MRSA in addition to streptococci, particularly in unresponsive or high-risk infections.
Basics description
Lymphadenitis refers to inflammation and enlargement of lymph nodes, most commonly as part of a systemic response to infection. Nodes become engorged with lymphocytes and macrophages and may be secondarily involved from infection in a distal extremity, producing painful, tender adenopathy proximally. Acute suppurative lymphadenitis may follow pharyngeal or skin infections and can progress to abscess formation.
Etiology
Lymphadenitis is most frequently caused by bacterial infection. The most common organisms in pyogenic lymphadenitis are Staphylococcus aureus, including community-associated methicillin-resistant S. aureus (CA-MRSA), and group A β-hemolytic Streptococcus. CA-MRSA risk factors include prior MRSA infection, household exposure, military service, incarceration, contact sports, injection drug use, and men who have sex with men. Cervical lymphadenitis usually originates from pharyngeal or periodontal infections and commonly involves streptococci and anaerobes. Axillary lymphadenitis is often caused by group A streptococcus. Nosocomial MRSA should be suspected in patients with recent hospitalization, surgery, dialysis, vascular catheters, recent antibiotic use, or unresponsive infection. In children, acute unilateral cervical suppurative lymphadenitis is most common in those younger than six years and is typically caused by S. aureus, group A streptococcus, or anaerobes.
Diagnosis signs and symptoms
Patients typically present with painful swelling and inflammation of affected lymph nodes, often in association with cellulitis or abscess if the cause is pyogenic. Axillary lymphadenitis may present with fever, axillary pain, and acute lymphedema of the arm or chest and may be associated with ipsilateral pleural effusion. History should include duration of lymphadenopathy, pain, fever, night sweats, weight loss, fatigue, sore throat, cough, occupational and animal exposures, sexual history, drug use, and travel. Physical examination should assess whether lymphadenopathy is localized or generalized, node size, tenderness, overlying skin changes, presence of skin lesions, splenomegaly, and involvement of supraclavicular or scalene nodes, which is always abnormal.
Essential workup
Acute regional lymphadenitis is usually a clinical diagnosis and often part of a broader infectious syndrome such as cellulitis. History and physical examination should focus on identifying an infectious source.
Diagnosis tests and interpretation
Laboratory testing is not always required. A CBC may show leukocytosis with left shift or be normal. Serologic testing for EBV, CMV, HIV, or other pathogens should be guided by clinical suspicion. Ultrasound or CT imaging is indicated when patients fail to improve with therapy or when suppuration is suspected. Percutaneous needle aspiration or surgical drainage should be considered if abscess formation occurs or if there is poor clinical response.
Differential diagnosis
The differential diagnosis includes common infections such as adenovirus, scarlet fever, cat scratch disease, fungal infections, and herpes zoster, as well as unusual infections including sporotrichosis, diphtheria, plague, anthrax, typhoid, rubella, and West Nile virus. Sexually transmitted infections, systemic infections such as HIV, infectious mononucleosis, toxoplasmosis, tuberculosis, hepatitis, and dengue should be considered. Noninfectious causes include drug reactions, malignancy, rheumatologic disorders, and pediatric-specific conditions such as Kawasaki disease and PFAPA syndrome.
Treatment
Initial management includes ensuring airway, breathing, and circulation stability. Treatment is directed at the underlying cause and should account for local resistance patterns, including CA-MRSA prevalence. Outpatient therapy typically lasts 7–10 days and includes limb elevation, moist heat, analgesics, and antibiotics. Abscesses require drainage with culture when possible. Skin-source infections are commonly treated with oral cephalexin plus trimethoprim–sulfamethoxazole or alternatives such as clindamycin or doxycycline. Pharyngeal or periodontal sources are treated with penicillin VK or alternatives such as clindamycin or amoxicillin–clavulanate. Inpatient therapy may require IV penicillin-based regimens with MRSA coverage using vancomycin or clindamycin when indicated.
Disposition and follow-up
Admission is indicated for toxic-appearing patients, those with immunosuppression or significant comorbidities, inability to tolerate oral therapy, or unreliable follow-up. Patients with mild infection who are nontoxic, can take oral antibiotics, and have reliable follow-up within 24–48 hours may be discharged. Failure to resolve promptly with antibiotics should prompt evaluation for malignancy or other serious causes, and lymph node biopsy may be indicated for persistent, large, or supraclavicular nodes.
Pearls and pitfalls
Staphylococcus species are the most common cause of acute regional pyogenic lymphadenitis. Empiric antibiotic therapy should include coverage for CA-MRSA in addition to streptococci, particularly in unresponsive or high-risk infections.
- Published on
Emergency and Acute Medicine – Lymphogranuloma Venereum
Basics description
Lymphogranuloma venereum (LGV) is a sexually transmitted infection characterized by an initial painless genital lesion followed by regional lymphatic spread. The disease progresses through primary, secondary, and tertiary stages and is responsive to appropriate antibacterial therapy in early phases. LGV is endemic in Southeast Asia, Latin America, parts of Africa, and the Caribbean, with increasing incidence among men who have sex with men. It is also known as struma, tropical bubo, or Nicolas–Favre–Durand disease.
Etiology
LGV is caused by Chlamydia trachomatis serotypes L1, L2, and L3.
Diagnosis signs and symptoms
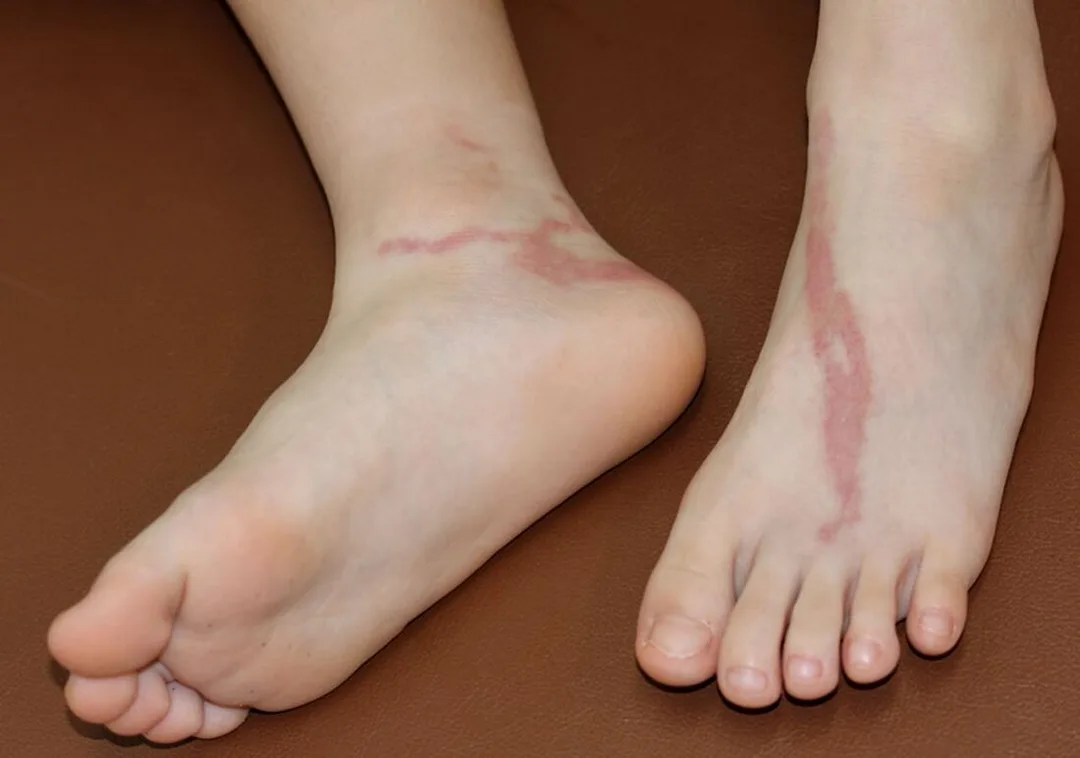
The primary stage occurs after an incubation period of 3–30 days and presents as a painless papule, pustule, vesicle, or ulcer in the anogenital region. These lesions are transient, often last only a few days, and are frequently unnoticed. The secondary stage develops 1–3 weeks later with systemic symptoms such as fever, malaise, and myalgias, along with tender inguinal lymphadenopathy that may be unilateral or bilateral. Large, fluctuant lymph nodes (buboes) can ulcerate and drain purulent material. Proctitis is common in anal-receptive patients and may cause rectal bleeding, tenesmus, and constipation. The tertiary stage occurs in untreated disease and results in chronic inflammatory changes including proctocolitis, strictures, fistulae, and elephantiasis of the genitalia or lower extremities, mimicking inflammatory bowel disease.
Physical examination
Findings in the primary stage include a painless anogenital lesion. During the secondary stage, tender inguinal or femoral lymphadenopathy is typical, with buboes forming in up to two-thirds of cases. The “groove sign,” caused by lymph node enlargement above and below the inguinal ligament, may be seen. Anal-receptive patients may demonstrate signs of hemorrhagic proctocolitis. Advanced tertiary disease shows chronic inflammatory damage with strictures, fistulae, and elephantiasis.
Diagnosis tests and interpretation
Routine chlamydia nucleic acid amplification tests do not differentiate LGV strains. Diagnosis is based on clinical suspicion, epidemiologic context, and serologic testing. Complement fixation titers greater than 1:64 support the diagnosis. False-positive VDRL tests may occur. Bubo aspiration is specific but rarely practical and is not routinely required.
Differential diagnosis
Conditions to consider include genital herpes, syphilis, chancroid, and granuloma inguinale. Compared with LGV, syphilis typically causes nontender lymphadenopathy with a longer incubation period, while chancroid presents with painful ulcers and granuloma inguinale causes painless, friable lesions that bleed easily.
Treatment
No prehospital or emergency stabilization is typically required. Large, fluctuant buboes may require aspiration or drainage to reduce pain and scarring. First-line therapy is doxycycline 100 mg orally twice daily for 21 days. Alternative regimens include azithromycin 1 g orally weekly for 3 weeks or erythromycin 500 mg orally four times daily for 3 weeks. In pregnancy or during lactation, erythromycin is the recommended treatment.
Disposition and follow-up
Hospitalization is rarely necessary and is reserved for patients with severe systemic illness or complications. Most immunocompetent patients without systemic involvement can be discharged with outpatient management. Sexual partners within the previous 60 days should be tested and treated with appropriate antichlamydial therapy. Follow-up is essential to confirm cure, particularly in cases of rectal involvement, which may require retreatment.
Pearls and pitfalls
LGV should be suspected in at-risk populations, particularly men who have sex with men presenting with inguinal lymphadenopathy or proctitis. Early recognition and treatment are critical to prevent progression to tertiary disease, which may cause irreversible tissue damage and is less responsive to antibiotics alone.
Basics description
Lymphogranuloma venereum (LGV) is a sexually transmitted infection characterized by an initial painless genital lesion followed by regional lymphatic spread. The disease progresses through primary, secondary, and tertiary stages and is responsive to appropriate antibacterial therapy in early phases. LGV is endemic in Southeast Asia, Latin America, parts of Africa, and the Caribbean, with increasing incidence among men who have sex with men. It is also known as struma, tropical bubo, or Nicolas–Favre–Durand disease.
Etiology
LGV is caused by Chlamydia trachomatis serotypes L1, L2, and L3.
Diagnosis signs and symptoms
The primary stage occurs after an incubation period of 3–30 days and presents as a painless papule, pustule, vesicle, or ulcer in the anogenital region. These lesions are transient, often last only a few days, and are frequently unnoticed. The secondary stage develops 1–3 weeks later with systemic symptoms such as fever, malaise, and myalgias, along with tender inguinal lymphadenopathy that may be unilateral or bilateral. Large, fluctuant lymph nodes (buboes) can ulcerate and drain purulent material. Proctitis is common in anal-receptive patients and may cause rectal bleeding, tenesmus, and constipation. The tertiary stage occurs in untreated disease and results in chronic inflammatory changes including proctocolitis, strictures, fistulae, and elephantiasis of the genitalia or lower extremities, mimicking inflammatory bowel disease.
Physical examination
Findings in the primary stage include a painless anogenital lesion. During the secondary stage, tender inguinal or femoral lymphadenopathy is typical, with buboes forming in up to two-thirds of cases. The “groove sign,” caused by lymph node enlargement above and below the inguinal ligament, may be seen. Anal-receptive patients may demonstrate signs of hemorrhagic proctocolitis. Advanced tertiary disease shows chronic inflammatory damage with strictures, fistulae, and elephantiasis.
Diagnosis tests and interpretation
Routine chlamydia nucleic acid amplification tests do not differentiate LGV strains. Diagnosis is based on clinical suspicion, epidemiologic context, and serologic testing. Complement fixation titers greater than 1:64 support the diagnosis. False-positive VDRL tests may occur. Bubo aspiration is specific but rarely practical and is not routinely required.
Differential diagnosis
Conditions to consider include genital herpes, syphilis, chancroid, and granuloma inguinale. Compared with LGV, syphilis typically causes nontender lymphadenopathy with a longer incubation period, while chancroid presents with painful ulcers and granuloma inguinale causes painless, friable lesions that bleed easily.
Treatment
No prehospital or emergency stabilization is typically required. Large, fluctuant buboes may require aspiration or drainage to reduce pain and scarring. First-line therapy is doxycycline 100 mg orally twice daily for 21 days. Alternative regimens include azithromycin 1 g orally weekly for 3 weeks or erythromycin 500 mg orally four times daily for 3 weeks. In pregnancy or during lactation, erythromycin is the recommended treatment.
Disposition and follow-up
Hospitalization is rarely necessary and is reserved for patients with severe systemic illness or complications. Most immunocompetent patients without systemic involvement can be discharged with outpatient management. Sexual partners within the previous 60 days should be tested and treated with appropriate antichlamydial therapy. Follow-up is essential to confirm cure, particularly in cases of rectal involvement, which may require retreatment.
Pearls and pitfalls
LGV should be suspected in at-risk populations, particularly men who have sex with men presenting with inguinal lymphadenopathy or proctitis. Early recognition and treatment are critical to prevent progression to tertiary disease, which may cause irreversible tissue damage and is less responsive to antibiotics alone.
- Published on
Emergency and Acute Medicine – Lymphangitis
Basics description
Lymphangitis is an infection of the lymphatic vessels that drain an area of inflammation or infection. Histologically, lymphatic channels become dilated and filled with lymphocytes and histiocytes. Inflammation often extends into surrounding perilymphatic tissues and may progress to cellulitis or abscess formation if untreated.
Etiology
Acute lymphangitis is most commonly caused by bacterial infection, particularly group A β-hemolytic Streptococcus. Less common causes include other streptococcal species and Staphylococcus aureus, including community-associated methicillin-resistant S. aureus (CA-MRSA). CA-MRSA risk factors include prior MRSA infection, close household contact, military service, incarceration, contact sports, injection drug use, and men who have sex with men. CA-MRSA is now prevalent enough to warrant empiric coverage in many settings, especially in unresponsive or recurrent infections. Other causes include Pasteurella multocida from animal bites, Spirillum minus in rat-bite fever, and Wuchereria bancrofti in filariasis, particularly in immigrants from endemic regions. Chronic lymphangitis is usually due to fungal, mycobacterial, or parasitic infections, most commonly Sporothrix schenckii, acquired through gardening or farming injuries, and Mycobacterium marinum, associated with fish tanks and swimming pools.
Diagnosis signs and symptoms
Acute lymphangitis presents with warm, tender, erythematous streaks extending proximally from a primary site of infection toward regional lymph nodes. Associated findings include painful lymphadenopathy, peripheral edema of the involved extremity, and systemic symptoms such as fever, rigors, tachycardia, and headache. Chronic nodular lymphangitis typically presents with painless erythematous nodules, chancriform ulcers, or wart-like lesions at the inoculation site, with possible progression along lymphatic channels and minimal systemic symptoms.
Essential workup
Lymphangitis is primarily a clinical diagnosis based on history and physical examination, with attention directed toward identifying the source of infection.
Diagnosis tests and interpretation
Laboratory testing is often unnecessary, though leukocytosis may be present. Gram stain and culture of aspirate or biopsy from the most inflamed area are recommended in cases of treatment failure or when resistant organisms such as MRSA are suspected. Culture confirmation is essential when sporotrichosis or M. marinum infection is considered. Blood cultures may be positive in severe cases. Imaging is rarely required but plain radiographs may help identify abscesses, subcutaneous gas, or foreign bodies, and Doppler ultrasound may be useful to exclude deep venous thrombosis.
Differential diagnosis
The differential diagnosis includes superficial or deep thrombophlebitis, which typically lacks an initial infectious focus or regional lymphadenopathy, IV line infiltration, vaccine-related reactions, and phytophotodermatitis, which can cause linear inflammatory skin changes mimicking lymphangitis.
Treatment
Initial management focuses on stabilizing septic patients with airway support and resuscitation as needed. Antimicrobial therapy should be initiated promptly, with the first dose given in the emergency department. Treatment typically lasts 7–10 days and includes limb elevation, moist heat, and antibiotics guided by local resistance patterns. Empiric outpatient therapy often includes oral cephalexin plus trimethoprim–sulfamethoxazole to cover CA-MRSA, with alternatives such as clindamycin or doxycycline. Inpatient therapy may require IV nafcillin or equivalent agents. Animal bite–associated lymphangitis is treated with IV ampicillin–sulbactam. Chronic infections require organism-specific therapy, such as itraconazole or potassium iodide for sporotrichosis and combination antimycobacterial therapy for M. marinum.
Disposition and follow-up
Admission is indicated for toxic-appearing patients, those with immunosuppression, significant comorbidities, inability to tolerate oral therapy, or unreliable follow-up. Patients with mild disease who are clinically stable and able to take oral antibiotics may be discharged with close follow-up within 24–48 hours. Marking the borders of erythema before discharge helps assess treatment response.
Pearls and pitfalls
Empiric antibiotic therapy should include coverage for CA-MRSA in addition to streptococcal species. Failure to recognize resistant organisms or chronic infectious causes can lead to treatment failure and progression of disease.
Basics description
Lymphangitis is an infection of the lymphatic vessels that drain an area of inflammation or infection. Histologically, lymphatic channels become dilated and filled with lymphocytes and histiocytes. Inflammation often extends into surrounding perilymphatic tissues and may progress to cellulitis or abscess formation if untreated.
Etiology
Acute lymphangitis is most commonly caused by bacterial infection, particularly group A β-hemolytic Streptococcus. Less common causes include other streptococcal species and Staphylococcus aureus, including community-associated methicillin-resistant S. aureus (CA-MRSA). CA-MRSA risk factors include prior MRSA infection, close household contact, military service, incarceration, contact sports, injection drug use, and men who have sex with men. CA-MRSA is now prevalent enough to warrant empiric coverage in many settings, especially in unresponsive or recurrent infections. Other causes include Pasteurella multocida from animal bites, Spirillum minus in rat-bite fever, and Wuchereria bancrofti in filariasis, particularly in immigrants from endemic regions. Chronic lymphangitis is usually due to fungal, mycobacterial, or parasitic infections, most commonly Sporothrix schenckii, acquired through gardening or farming injuries, and Mycobacterium marinum, associated with fish tanks and swimming pools.
Diagnosis signs and symptoms
Acute lymphangitis presents with warm, tender, erythematous streaks extending proximally from a primary site of infection toward regional lymph nodes. Associated findings include painful lymphadenopathy, peripheral edema of the involved extremity, and systemic symptoms such as fever, rigors, tachycardia, and headache. Chronic nodular lymphangitis typically presents with painless erythematous nodules, chancriform ulcers, or wart-like lesions at the inoculation site, with possible progression along lymphatic channels and minimal systemic symptoms.
Essential workup
Lymphangitis is primarily a clinical diagnosis based on history and physical examination, with attention directed toward identifying the source of infection.
Diagnosis tests and interpretation
Laboratory testing is often unnecessary, though leukocytosis may be present. Gram stain and culture of aspirate or biopsy from the most inflamed area are recommended in cases of treatment failure or when resistant organisms such as MRSA are suspected. Culture confirmation is essential when sporotrichosis or M. marinum infection is considered. Blood cultures may be positive in severe cases. Imaging is rarely required but plain radiographs may help identify abscesses, subcutaneous gas, or foreign bodies, and Doppler ultrasound may be useful to exclude deep venous thrombosis.
Differential diagnosis
The differential diagnosis includes superficial or deep thrombophlebitis, which typically lacks an initial infectious focus or regional lymphadenopathy, IV line infiltration, vaccine-related reactions, and phytophotodermatitis, which can cause linear inflammatory skin changes mimicking lymphangitis.
Treatment
Initial management focuses on stabilizing septic patients with airway support and resuscitation as needed. Antimicrobial therapy should be initiated promptly, with the first dose given in the emergency department. Treatment typically lasts 7–10 days and includes limb elevation, moist heat, and antibiotics guided by local resistance patterns. Empiric outpatient therapy often includes oral cephalexin plus trimethoprim–sulfamethoxazole to cover CA-MRSA, with alternatives such as clindamycin or doxycycline. Inpatient therapy may require IV nafcillin or equivalent agents. Animal bite–associated lymphangitis is treated with IV ampicillin–sulbactam. Chronic infections require organism-specific therapy, such as itraconazole or potassium iodide for sporotrichosis and combination antimycobacterial therapy for M. marinum.
Disposition and follow-up
Admission is indicated for toxic-appearing patients, those with immunosuppression, significant comorbidities, inability to tolerate oral therapy, or unreliable follow-up. Patients with mild disease who are clinically stable and able to take oral antibiotics may be discharged with close follow-up within 24–48 hours. Marking the borders of erythema before discharge helps assess treatment response.
Pearls and pitfalls
Empiric antibiotic therapy should include coverage for CA-MRSA in addition to streptococcal species. Failure to recognize resistant organisms or chronic infectious causes can lead to treatment failure and progression of disease.
- Published on
KembaraXtra-Medicine – Intestinal Malrotation
Intestinal malrotation is a congenital condition resulting from incomplete rotation and fixation of the intestine during embryogenesis, specifically during the transition from the extracolonic position around the tenth week of gestation. It is commonly associated with heterotaxia syndromes and frequently occurs alongside other congenital anomalies. Gastrointestinal associations include duodenal stenosis or atresia, duodenal web, Meckel diverticulum, intussusception, gastroesophageal reflux, omphalocele, gastroschisis, congenital diaphragmatic hernia, abdominal wall defects, and Hirschsprung disease. Congenital cardiac anomalies are present in approximately 27% of patients and significantly increase morbidity.
The underlying pathology involves abnormal positioning of the duodenojejunal junction, which remains to the right of the midline, and malposition of the cecum, which is often located in the upper left abdomen with abnormal mesenteric attachments. This abnormal anatomy predisposes patients to bowel obstruction and volvulus. Midgut volvulus is a serious complication that occurs when the small intestine twists around the superior mesenteric artery and vein, leading to vascular compromise and potential bowel ischemia. Malrotation is found in combination with other congenital anomalies in up to 70% of cases, particularly involving the cardiac, esophageal, urinary, and anorectal systems.
Malrotation occurs in approximately 1 in 500 live births. It is more common in males, with a male-to-female ratio of 2:1 in neonates. About 75% of cases are diagnosed during the newborn period, and 90% are identified by one year of age, although presentation can occur in adulthood. Mortality in infants can be as high as 24%, and the presence of necrotic bowel at surgery increases mortality risk significantly.
Clinical presentation varies by age. Neonates typically present with bilious vomiting, abdominal distention, bloody stools, constipation or obstipation, feeding difficulties, and poor weight gain. Infants and children older than one year may present with abdominal pain followed by bilious emesis. Older children and adolescents often experience chronic vomiting, intermittent colicky abdominal pain, diarrhea, hematemesis, or constipation. Adults usually present with vague and nonspecific gastrointestinal symptoms. Notably, up to 75% of patients may have a normal physical examination at presentation. Severe cases may show signs of dehydration, metabolic acidosis, peritonitis, ischemic bowel, sepsis, or shock.
Diagnosis is suggested by clinical history and physical examination and is confirmed through imaging, most commonly contrast radiography. Laboratory evaluation includes complete blood count, venous blood gas, electrolytes, renal function tests, glucose, coagulation profile, lactate, and type and screen. Plain abdominal radiographs are diagnostic in fewer than 30% of cases but may show signs suggestive of volvulus, such as duodenal obstruction, gastric distention, paucity of distal bowel gas, generalized small-bowel distention, or a double-bubble sign. Upper gastrointestinal contrast studies are the diagnostic test of choice, with high sensitivity and accuracy, demonstrating abnormal position of the duodenojejunal junction, corkscrew appearance of the duodenum in volvulus, or right-sided jejunum. Contrast enema may help identify cecal position but has a significant false-negative rate. Ultrasound can demonstrate abnormal superior mesenteric artery–vein relationships and the classic whirlpool sign in volvulus, although a normal study does not exclude malrotation. CT imaging is generally reserved for adults.
Differential diagnosis depends on age and presentation. In early life, considerations include Hirschsprung disease, necrotizing enterocolitis, and intussusception. In children with acute abdominal pain and peritoneal signs, appendicitis and sepsis must be considered. Older children and adults with chronic or vague symptoms may be misdiagnosed with irritable bowel syndrome, peptic ulcer disease, biliary or pancreatic disorders, or psychiatric conditions.
Management of malrotation, particularly with midgut volvulus, is a surgical emergency. Initial stabilization focuses on airway, breathing, and circulation, with rapid intravenous fluid resuscitation to correct hypovolemia and metabolic acidosis. Broad-spectrum antibiotics are initiated if there are signs of sepsis or peritonitis. Nasogastric decompression and prompt surgical consultation are essential. Definitive treatment involves emergent surgical correction, including detorsion of the volvulus, restoration of intestinal perfusion, resection of necrotic bowel if present, and reassessment of bowel viability when necessary. Patients are kept nil per os and require close postoperative monitoring.
Hospital admission is indicated for acute abdomen, need for surgical intervention, significant dehydration, acidosis, sepsis, or shock. Discharge is uncommon and reserved for stable, asymptomatic patients with incidental findings after thorough pediatric surgical evaluation. Early recognition, rapid resuscitation, and timely referral to a facility with pediatric surgical expertise are critical to improving outcomes and reducing morbidity and mortality.
Intestinal malrotation is a congenital condition resulting from incomplete rotation and fixation of the intestine during embryogenesis, specifically during the transition from the extracolonic position around the tenth week of gestation. It is commonly associated with heterotaxia syndromes and frequently occurs alongside other congenital anomalies. Gastrointestinal associations include duodenal stenosis or atresia, duodenal web, Meckel diverticulum, intussusception, gastroesophageal reflux, omphalocele, gastroschisis, congenital diaphragmatic hernia, abdominal wall defects, and Hirschsprung disease. Congenital cardiac anomalies are present in approximately 27% of patients and significantly increase morbidity.
The underlying pathology involves abnormal positioning of the duodenojejunal junction, which remains to the right of the midline, and malposition of the cecum, which is often located in the upper left abdomen with abnormal mesenteric attachments. This abnormal anatomy predisposes patients to bowel obstruction and volvulus. Midgut volvulus is a serious complication that occurs when the small intestine twists around the superior mesenteric artery and vein, leading to vascular compromise and potential bowel ischemia. Malrotation is found in combination with other congenital anomalies in up to 70% of cases, particularly involving the cardiac, esophageal, urinary, and anorectal systems.
Malrotation occurs in approximately 1 in 500 live births. It is more common in males, with a male-to-female ratio of 2:1 in neonates. About 75% of cases are diagnosed during the newborn period, and 90% are identified by one year of age, although presentation can occur in adulthood. Mortality in infants can be as high as 24%, and the presence of necrotic bowel at surgery increases mortality risk significantly.
Clinical presentation varies by age. Neonates typically present with bilious vomiting, abdominal distention, bloody stools, constipation or obstipation, feeding difficulties, and poor weight gain. Infants and children older than one year may present with abdominal pain followed by bilious emesis. Older children and adolescents often experience chronic vomiting, intermittent colicky abdominal pain, diarrhea, hematemesis, or constipation. Adults usually present with vague and nonspecific gastrointestinal symptoms. Notably, up to 75% of patients may have a normal physical examination at presentation. Severe cases may show signs of dehydration, metabolic acidosis, peritonitis, ischemic bowel, sepsis, or shock.
Diagnosis is suggested by clinical history and physical examination and is confirmed through imaging, most commonly contrast radiography. Laboratory evaluation includes complete blood count, venous blood gas, electrolytes, renal function tests, glucose, coagulation profile, lactate, and type and screen. Plain abdominal radiographs are diagnostic in fewer than 30% of cases but may show signs suggestive of volvulus, such as duodenal obstruction, gastric distention, paucity of distal bowel gas, generalized small-bowel distention, or a double-bubble sign. Upper gastrointestinal contrast studies are the diagnostic test of choice, with high sensitivity and accuracy, demonstrating abnormal position of the duodenojejunal junction, corkscrew appearance of the duodenum in volvulus, or right-sided jejunum. Contrast enema may help identify cecal position but has a significant false-negative rate. Ultrasound can demonstrate abnormal superior mesenteric artery–vein relationships and the classic whirlpool sign in volvulus, although a normal study does not exclude malrotation. CT imaging is generally reserved for adults.
Differential diagnosis depends on age and presentation. In early life, considerations include Hirschsprung disease, necrotizing enterocolitis, and intussusception. In children with acute abdominal pain and peritoneal signs, appendicitis and sepsis must be considered. Older children and adults with chronic or vague symptoms may be misdiagnosed with irritable bowel syndrome, peptic ulcer disease, biliary or pancreatic disorders, or psychiatric conditions.
Management of malrotation, particularly with midgut volvulus, is a surgical emergency. Initial stabilization focuses on airway, breathing, and circulation, with rapid intravenous fluid resuscitation to correct hypovolemia and metabolic acidosis. Broad-spectrum antibiotics are initiated if there are signs of sepsis or peritonitis. Nasogastric decompression and prompt surgical consultation are essential. Definitive treatment involves emergent surgical correction, including detorsion of the volvulus, restoration of intestinal perfusion, resection of necrotic bowel if present, and reassessment of bowel viability when necessary. Patients are kept nil per os and require close postoperative monitoring.
Hospital admission is indicated for acute abdomen, need for surgical intervention, significant dehydration, acidosis, sepsis, or shock. Discharge is uncommon and reserved for stable, asymptomatic patients with incidental findings after thorough pediatric surgical evaluation. Early recognition, rapid resuscitation, and timely referral to a facility with pediatric surgical expertise are critical to improving outcomes and reducing morbidity and mortality.
- Published on
KembaraXtra-Medicine – Autosomal Dominant Polycystic Kidney Disease (ADPKD)
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited form of kidney disease and represents a systemic genetic disorder. It is caused predominantly by sequence variations in the PKD1 and PKD2 genes, which account for approximately 93% of cases. These genetic changes result in progressive formation and enlargement of fluid-filled cysts, mainly in the kidneys, but also in other organs such as the liver and pancreas. In addition to renal involvement, ADPKD is associated with gastrointestinal, connective tissue, and cardiovascular abnormalities.
ADPKD follows an autosomal dominant inheritance pattern, meaning each child of an affected parent has a 50% chance of inheriting the disease. It affects individuals of all ethnic groups worldwide and is the most common hereditary cause of chronic kidney disease and end-stage renal disease. Most genetically confirmed cases are due to PKD1 variants on chromosome 16, while PKD2 variants on chromosome 4 account for a smaller proportion and are usually associated with milder disease and later progression. A minority of patients have variants in other genes such as GANAB, DNAJB11, ALG8, ALG9, ALG5, and IFT140, which often produce atypical or milder phenotypes.
The disease mechanism involves abnormal function of polycystin proteins, which play essential roles in epithelial cell differentiation, intracellular calcium signaling, and primary ciliary function. Disruption of these pathways leads to cyst formation, epithelial proliferation, and progressive kidney enlargement. Cyst burden and total kidney volume increase over time and are key drivers of disease progression and loss of kidney function.
Clinically, ADPKD is often asymptomatic in the first two to three decades of life. Many patients are diagnosed through family screening or incidental imaging. As cyst burden increases, patients may develop flank pain, hematuria, kidney stones, urinary tract infections, polyuria, nocturia, and hypertension. Gross hematuria commonly results from cyst rupture and is associated with larger kidney size. Proteinuria is usually mild but correlates with more advanced disease.
Hypertension is one of the earliest and most common manifestations of ADPKD and frequently occurs before a decline in kidney function. It is primarily driven by activation of the intrarenal renin–angiotensin–aldosterone system and strongly correlates with cyst burden. Cardiac involvement may include valvular abnormalities such as mitral valve prolapse and aortic regurgitation, contributing to cardiovascular morbidity.
Extrarenal manifestations are common and clinically significant. Liver cysts are the most frequent and increase in prevalence with age, often becoming apparent by early adulthood. Although usually asymptomatic, hepatic cysts can cause pain, infection, bleeding, and biliary obstruction. Intracranial aneurysms occur more frequently in ADPKD than in the general population and carry a risk of rupture, particularly in patients with a family history of aneurysmal bleeding. Other manifestations include pancreatic cysts, colonic diverticula, abdominal and inguinal hernias, and seminal vesicle cysts in men.
Disease progression varies widely. Patients with PKD1 variants generally experience more rapid progression than those with PKD2 variants. Truncating PKD1 mutations are associated with earlier onset of end-stage renal disease. Risk factors for faster progression include male sex, early-onset hypertension, early gross hematuria, proteinuria, and greater total kidney volume. Imaging-based classifications, such as the Mayo Clinic Imaging Classification, use height-adjusted total kidney volume to stratify patients according to risk of progression.
Diagnosis is primarily based on imaging. Renal ultrasound is the most commonly used screening and diagnostic tool due to its availability and lack of radiation. CT and MRI are more sensitive and are often reserved for evaluating complications or measuring total kidney volume. In individuals with a family history, age-specific cyst number criteria are used to establish the diagnosis. Genetic testing may be considered when imaging findings are equivocal or when a definitive diagnosis is required for family planning or living kidney donation.
Management of ADPKD focuses on slowing disease progression, managing complications, and reducing cardiovascular risk. Nonpharmacologic measures include dietary sodium restriction and adequate hydration, particularly in patients with preserved kidney function. Strict blood pressure control, preferably with ACE inhibitors or angiotensin receptor blockers, is central to therapy. Acute complications such as hematuria, infections, and nephrolithiasis are treated supportively and with targeted therapies.
Tolvaptan, a vasopressin V2 receptor antagonist, is the first disease-modifying therapy approved for ADPKD in adults at risk for rapid progression. Clinical trials have demonstrated its ability to slow kidney growth and decline in kidney function, particularly in patients with high cyst burden. Its use requires careful monitoring due to risks of polyuria, dehydration, hypernatremia, and hepatotoxicity. Other investigational and adjunctive therapies are under study, but none have yet shown consistent benefit comparable to tolvaptan.
Patients with ADPKD should be referred early to a nephrologist for longitudinal care and risk stratification. Genetic counseling is recommended for affected individuals and families, especially when considering pregnancy or screening of at-risk relatives. Early recognition, individualized risk assessment, and comprehensive management are essential to improving long-term outcomes in ADPKD.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited form of kidney disease and represents a systemic genetic disorder. It is caused predominantly by sequence variations in the PKD1 and PKD2 genes, which account for approximately 93% of cases. These genetic changes result in progressive formation and enlargement of fluid-filled cysts, mainly in the kidneys, but also in other organs such as the liver and pancreas. In addition to renal involvement, ADPKD is associated with gastrointestinal, connective tissue, and cardiovascular abnormalities.
ADPKD follows an autosomal dominant inheritance pattern, meaning each child of an affected parent has a 50% chance of inheriting the disease. It affects individuals of all ethnic groups worldwide and is the most common hereditary cause of chronic kidney disease and end-stage renal disease. Most genetically confirmed cases are due to PKD1 variants on chromosome 16, while PKD2 variants on chromosome 4 account for a smaller proportion and are usually associated with milder disease and later progression. A minority of patients have variants in other genes such as GANAB, DNAJB11, ALG8, ALG9, ALG5, and IFT140, which often produce atypical or milder phenotypes.
The disease mechanism involves abnormal function of polycystin proteins, which play essential roles in epithelial cell differentiation, intracellular calcium signaling, and primary ciliary function. Disruption of these pathways leads to cyst formation, epithelial proliferation, and progressive kidney enlargement. Cyst burden and total kidney volume increase over time and are key drivers of disease progression and loss of kidney function.
Clinically, ADPKD is often asymptomatic in the first two to three decades of life. Many patients are diagnosed through family screening or incidental imaging. As cyst burden increases, patients may develop flank pain, hematuria, kidney stones, urinary tract infections, polyuria, nocturia, and hypertension. Gross hematuria commonly results from cyst rupture and is associated with larger kidney size. Proteinuria is usually mild but correlates with more advanced disease.
Hypertension is one of the earliest and most common manifestations of ADPKD and frequently occurs before a decline in kidney function. It is primarily driven by activation of the intrarenal renin–angiotensin–aldosterone system and strongly correlates with cyst burden. Cardiac involvement may include valvular abnormalities such as mitral valve prolapse and aortic regurgitation, contributing to cardiovascular morbidity.
Extrarenal manifestations are common and clinically significant. Liver cysts are the most frequent and increase in prevalence with age, often becoming apparent by early adulthood. Although usually asymptomatic, hepatic cysts can cause pain, infection, bleeding, and biliary obstruction. Intracranial aneurysms occur more frequently in ADPKD than in the general population and carry a risk of rupture, particularly in patients with a family history of aneurysmal bleeding. Other manifestations include pancreatic cysts, colonic diverticula, abdominal and inguinal hernias, and seminal vesicle cysts in men.
Disease progression varies widely. Patients with PKD1 variants generally experience more rapid progression than those with PKD2 variants. Truncating PKD1 mutations are associated with earlier onset of end-stage renal disease. Risk factors for faster progression include male sex, early-onset hypertension, early gross hematuria, proteinuria, and greater total kidney volume. Imaging-based classifications, such as the Mayo Clinic Imaging Classification, use height-adjusted total kidney volume to stratify patients according to risk of progression.
Diagnosis is primarily based on imaging. Renal ultrasound is the most commonly used screening and diagnostic tool due to its availability and lack of radiation. CT and MRI are more sensitive and are often reserved for evaluating complications or measuring total kidney volume. In individuals with a family history, age-specific cyst number criteria are used to establish the diagnosis. Genetic testing may be considered when imaging findings are equivocal or when a definitive diagnosis is required for family planning or living kidney donation.
Management of ADPKD focuses on slowing disease progression, managing complications, and reducing cardiovascular risk. Nonpharmacologic measures include dietary sodium restriction and adequate hydration, particularly in patients with preserved kidney function. Strict blood pressure control, preferably with ACE inhibitors or angiotensin receptor blockers, is central to therapy. Acute complications such as hematuria, infections, and nephrolithiasis are treated supportively and with targeted therapies.
Tolvaptan, a vasopressin V2 receptor antagonist, is the first disease-modifying therapy approved for ADPKD in adults at risk for rapid progression. Clinical trials have demonstrated its ability to slow kidney growth and decline in kidney function, particularly in patients with high cyst burden. Its use requires careful monitoring due to risks of polyuria, dehydration, hypernatremia, and hepatotoxicity. Other investigational and adjunctive therapies are under study, but none have yet shown consistent benefit comparable to tolvaptan.
Patients with ADPKD should be referred early to a nephrologist for longitudinal care and risk stratification. Genetic counseling is recommended for affected individuals and families, especially when considering pregnancy or screening of at-risk relatives. Early recognition, individualized risk assessment, and comprehensive management are essential to improving long-term outcomes in ADPKD.

- Published on
KembaraXtra-Medicine – Autoimmune Hemolytic Anemia
Autoimmune hemolytic anemia (AIHA) is a condition in which autoantibodies and/or complement bind to red blood cells (RBCs), leading to their premature destruction. About half of cases are primary (idiopathic), while the rest are secondary to underlying diseases or drugs. AIHA is commonly categorized into warm antibody–mediated disease (usually IgG, reacting best at 37°C), cold antibody–mediated disease (typically IgM with complement), and drug-induced immune hemolysis.
AIHA is uncommon, with an annual incidence of about 1 to 3 cases per 100,000 people and an estimated mortality around 10%. It is reported more often in women younger than 50 years. Patients most commonly present with fatigue and dyspnea. Pallor and jaundice may be present, and tachycardia with a flow murmur can occur when anemia is significant. Intravascular hemolysis may cause dark urine and back pain. Hepatomegaly or lymphadenopathy suggests a lymphoproliferative disorder or malignancy, while splenomegaly may suggest hypersplenism. The course is often chronic with relapses.
Warm AIHA is usually mediated by IgG antibodies and may be idiopathic or associated with leukemia/lymphoma, thymoma, myeloma, viral infections, babesiosis, and collagen-vascular diseases. Cold AIHA is commonly IgM- and complement-mediated and may be idiopathic or linked to infections, lymphoma, or cold agglutinin disease. Drug-induced AIHA can occur through different immune mechanisms, including antibodies directed against RBC antigens (e.g., methyldopa), antibodies against an RBC–drug complex (hapten type, e.g., penicillin), or immune complex mechanisms (e.g., quinidine).
The differential diagnosis includes non-immune causes of hemolysis such as microangiopathic hemolytic anemia (e.g., TTP/HUS), hypersplenism, prosthetic valve hemolysis, infections, toxins, and inherited causes such as membrane disorders (e.g., hereditary spherocytosis), hemoglobinopathies, and enzyme deficiencies (e.g., G6PD). Chronic lymphocytic leukemia (CLL) can cause a positive direct antiglobulin test (DAT) without true AIHA, so labs and clinical evidence of hemolysis are important.
Evaluation focuses on confirming hemolysis and establishing immune causation. Typical hemolysis findings include reticulocytosis (or reticulocytopenia if marrow suppression is present), low haptoglobin, elevated indirect bilirubin, and elevated LDH. Initial tests include CBC, reticulocyte count, bilirubin/LDH, and a peripheral smear (spherocytes are typical in warm AIHA). The key diagnostic test is the direct antiglobulin test (DAT/Coombs), performed initially with a polyspecific reagent to detect IgG and/or complement on RBCs. A positive DAT supports AIHA: IgG ± C3d suggests warm AIHA, while C3d alone suggests cold AIHA. Additional evaluation may include hepatitis serologies, HIV testing, ANA, urine testing for hemoglobinuria/hemosiderinuria, and imaging (such as CT chest/abdomen/pelvis) if an underlying lymphoproliferative disorder is suspected.
Management includes stopping any offending drug when drug-induced disease is possible. Severe, life-threatening cases may rarely require plasmapheresis or exchange transfusion. Patients with cold antibody AIHA should avoid cold exposure. Warm AIHA is typically treated first with prednisone 1 to 2 mg/kg/day, with tapering after response. Some evidence supports upfront combination therapy with steroids plus rituximab to improve outcomes and durability of response. For relapse or steroid-refractory disease, rituximab is commonly used and has high response rates, with a substantial portion maintaining remission years later. Splenectomy is now usually reserved for cases refractory to both steroids and rituximab, and further options include immunosuppressive agents such as mycophenolate, cyclosporine, cyclophosphamide, IVIG, and sometimes danazol as an adjunct.
Cold AIHA generally responds poorly to corticosteroids, and treating underlying infection or lymphoproliferative disease is important. In primary cold agglutinin disease, rituximab-based regimens (often with bendamustine in fit patients) are commonly used, while rituximab alone may be used in older or frailer patients. IVIG or plasmapheresis can be used temporarily in severe cases while waiting for a durable therapy to work. Complement-directed therapies may be considered in severe disease; sutimlimab (targeting C1s) has been shown to rapidly reduce hemolysis and improve hemoglobin and fatigue in cold agglutinin disease. Splenectomy is generally ineffective in cold AIHA because clearance occurs mainly in the liver.
Prognosis is generally good unless AIHA is driven by a serious underlying condition such as leukemia or myeloma. Hematology referral is recommended for all AIHA cases, and surgical referral may be needed if splenectomy is being considered for refractory warm AIHA.
Autoimmune hemolytic anemia (AIHA) is a condition in which autoantibodies and/or complement bind to red blood cells (RBCs), leading to their premature destruction. About half of cases are primary (idiopathic), while the rest are secondary to underlying diseases or drugs. AIHA is commonly categorized into warm antibody–mediated disease (usually IgG, reacting best at 37°C), cold antibody–mediated disease (typically IgM with complement), and drug-induced immune hemolysis.
AIHA is uncommon, with an annual incidence of about 1 to 3 cases per 100,000 people and an estimated mortality around 10%. It is reported more often in women younger than 50 years. Patients most commonly present with fatigue and dyspnea. Pallor and jaundice may be present, and tachycardia with a flow murmur can occur when anemia is significant. Intravascular hemolysis may cause dark urine and back pain. Hepatomegaly or lymphadenopathy suggests a lymphoproliferative disorder or malignancy, while splenomegaly may suggest hypersplenism. The course is often chronic with relapses.
Warm AIHA is usually mediated by IgG antibodies and may be idiopathic or associated with leukemia/lymphoma, thymoma, myeloma, viral infections, babesiosis, and collagen-vascular diseases. Cold AIHA is commonly IgM- and complement-mediated and may be idiopathic or linked to infections, lymphoma, or cold agglutinin disease. Drug-induced AIHA can occur through different immune mechanisms, including antibodies directed against RBC antigens (e.g., methyldopa), antibodies against an RBC–drug complex (hapten type, e.g., penicillin), or immune complex mechanisms (e.g., quinidine).
The differential diagnosis includes non-immune causes of hemolysis such as microangiopathic hemolytic anemia (e.g., TTP/HUS), hypersplenism, prosthetic valve hemolysis, infections, toxins, and inherited causes such as membrane disorders (e.g., hereditary spherocytosis), hemoglobinopathies, and enzyme deficiencies (e.g., G6PD). Chronic lymphocytic leukemia (CLL) can cause a positive direct antiglobulin test (DAT) without true AIHA, so labs and clinical evidence of hemolysis are important.
Evaluation focuses on confirming hemolysis and establishing immune causation. Typical hemolysis findings include reticulocytosis (or reticulocytopenia if marrow suppression is present), low haptoglobin, elevated indirect bilirubin, and elevated LDH. Initial tests include CBC, reticulocyte count, bilirubin/LDH, and a peripheral smear (spherocytes are typical in warm AIHA). The key diagnostic test is the direct antiglobulin test (DAT/Coombs), performed initially with a polyspecific reagent to detect IgG and/or complement on RBCs. A positive DAT supports AIHA: IgG ± C3d suggests warm AIHA, while C3d alone suggests cold AIHA. Additional evaluation may include hepatitis serologies, HIV testing, ANA, urine testing for hemoglobinuria/hemosiderinuria, and imaging (such as CT chest/abdomen/pelvis) if an underlying lymphoproliferative disorder is suspected.
Management includes stopping any offending drug when drug-induced disease is possible. Severe, life-threatening cases may rarely require plasmapheresis or exchange transfusion. Patients with cold antibody AIHA should avoid cold exposure. Warm AIHA is typically treated first with prednisone 1 to 2 mg/kg/day, with tapering after response. Some evidence supports upfront combination therapy with steroids plus rituximab to improve outcomes and durability of response. For relapse or steroid-refractory disease, rituximab is commonly used and has high response rates, with a substantial portion maintaining remission years later. Splenectomy is now usually reserved for cases refractory to both steroids and rituximab, and further options include immunosuppressive agents such as mycophenolate, cyclosporine, cyclophosphamide, IVIG, and sometimes danazol as an adjunct.
Cold AIHA generally responds poorly to corticosteroids, and treating underlying infection or lymphoproliferative disease is important. In primary cold agglutinin disease, rituximab-based regimens (often with bendamustine in fit patients) are commonly used, while rituximab alone may be used in older or frailer patients. IVIG or plasmapheresis can be used temporarily in severe cases while waiting for a durable therapy to work. Complement-directed therapies may be considered in severe disease; sutimlimab (targeting C1s) has been shown to rapidly reduce hemolysis and improve hemoglobin and fatigue in cold agglutinin disease. Splenectomy is generally ineffective in cold AIHA because clearance occurs mainly in the liver.
Prognosis is generally good unless AIHA is driven by a serious underlying condition such as leukemia or myeloma. Hematology referral is recommended for all AIHA cases, and surgical referral may be needed if splenectomy is being considered for refractory warm AIHA.

- Published on
Emergency and Acute Medicine – Malaria
Basics description
Malaria is a protozoan infection transmitted through the bite of an infected female Anopheles mosquito. The incubation period is typically 8–16 days, although symptoms may be delayed for months depending on the species. The periodic nature of malaria symptoms reflects the parasite life cycle, which includes an exoerythrocytic phase in the liver followed by an erythrocytic phase in red blood cells. Fever corresponds to synchronous red blood cell lysis and release of merozoites into the circulation.
Pathophysiology and species differences
In the exoerythrocytic phase, sporozoites migrate to the liver and multiply into merozoites. These then enter the bloodstream and invade red blood cells, where replication leads to cell rupture every 48–72 hours. Plasmodium falciparum causes most severe disease and nearly all malaria-related deaths. It infects red blood cells of all ages, leading to profound hemolysis, anemia, and capillary obstruction with resultant end-organ hypoxia. Plasmodium vivax and Plasmodium ovale may cause relapsing disease due to dormant liver forms (hypnozoites), while Plasmodium malariae can persist at low levels in the bloodstream for decades.
Etiology and transmission
Transmission usually occurs via mosquito bite but may also result from blood transfusions or shared needles. Rare autochthonous transmission has been reported in North America due to the presence of Anopheles mosquitoes. Post-traumatic immunosuppression may precipitate relapse in individuals previously exposed in endemic regions. Children with sickle cell trait have partial protection, while pregnant patients—especially primigravidas—are at higher risk for severe disease.
Diagnosis signs and symptoms
Most patients become symptomatic within one year of exposure. Common symptoms include malaise, chills, fever greater than 38°C, myalgias, arthralgias, and orthostatic hypotension. The classic malaria paroxysm consists of chills followed by high fever and subsequent diaphoresis, although this pattern is uncommon in clinical practice. Hemolysis may lead to jaundice, dark urine (blackwater fever), anemia, and splenomegaly. Severe manifestations include cerebral malaria with headache, altered mental status, seizures, or coma; pulmonary edema; renal failure; abnormal bleeding; and circulatory collapse.
Essential workup and diagnostic testing
Diagnosis relies on oil immersion light microscopy of thick and thin Giemsa-stained blood smears, which demonstrate intraerythrocytic parasites. At least three negative smears over 48 hours are required to exclude malaria. Laboratory findings commonly include anemia, thrombocytopenia, leukocytopenia, elevated bilirubin and lactate dehydrogenase, electrolyte abnormalities, and evidence of renal or hepatic dysfunction. Chest radiography may reveal pulmonary edema. Advanced testing such as antigen detection assays or PCR can identify Plasmodium species and mixed infections. Lumbar puncture may be necessary to distinguish cerebral malaria from meningitis.
Differential diagnosis
The differential includes meningitis, encephalitis, sepsis, acute renal failure, viral hepatitis, acute hemolytic anemia, hypoglycemic coma, and heat stroke. In travelers or immigrants from endemic areas, malaria must be considered in any febrile illness.
Treatment and emergency management
Initial stabilization includes airway, breathing, and circulation management, isotonic fluid resuscitation for hypotension, active cooling for hyperthermia, and treatment of hypoglycemia or seizures as indicated. Antimalarial therapy depends on species, severity, and geographic resistance patterns, with artemisinin-based combination therapies recommended as first-line treatment worldwide. Severe P. falciparum infection requires intravenous artesunate. Supportive care for complications such as anemia, renal failure, and cerebral edema is critical.
Disposition and follow-up
ICU admission is indicated for severe P. falciparum malaria, high parasitemia, neurologic involvement, or inability to tolerate oral therapy. Patients with non–P. falciparum malaria who are clinically stable and able to take oral medications may be treated as outpatients with close follow-up.
Pearls and pitfalls
Malaria should always be considered in patients with fever and compatible exposure history, even months after travel. Fever patterns are unreliable, and early recognition is essential, as delays in treatment—especially with P. falciparum—are associated with high mortality.
Basics description
Malaria is a protozoan infection transmitted through the bite of an infected female Anopheles mosquito. The incubation period is typically 8–16 days, although symptoms may be delayed for months depending on the species. The periodic nature of malaria symptoms reflects the parasite life cycle, which includes an exoerythrocytic phase in the liver followed by an erythrocytic phase in red blood cells. Fever corresponds to synchronous red blood cell lysis and release of merozoites into the circulation.
Pathophysiology and species differences
In the exoerythrocytic phase, sporozoites migrate to the liver and multiply into merozoites. These then enter the bloodstream and invade red blood cells, where replication leads to cell rupture every 48–72 hours. Plasmodium falciparum causes most severe disease and nearly all malaria-related deaths. It infects red blood cells of all ages, leading to profound hemolysis, anemia, and capillary obstruction with resultant end-organ hypoxia. Plasmodium vivax and Plasmodium ovale may cause relapsing disease due to dormant liver forms (hypnozoites), while Plasmodium malariae can persist at low levels in the bloodstream for decades.
Etiology and transmission
Transmission usually occurs via mosquito bite but may also result from blood transfusions or shared needles. Rare autochthonous transmission has been reported in North America due to the presence of Anopheles mosquitoes. Post-traumatic immunosuppression may precipitate relapse in individuals previously exposed in endemic regions. Children with sickle cell trait have partial protection, while pregnant patients—especially primigravidas—are at higher risk for severe disease.
Diagnosis signs and symptoms
Most patients become symptomatic within one year of exposure. Common symptoms include malaise, chills, fever greater than 38°C, myalgias, arthralgias, and orthostatic hypotension. The classic malaria paroxysm consists of chills followed by high fever and subsequent diaphoresis, although this pattern is uncommon in clinical practice. Hemolysis may lead to jaundice, dark urine (blackwater fever), anemia, and splenomegaly. Severe manifestations include cerebral malaria with headache, altered mental status, seizures, or coma; pulmonary edema; renal failure; abnormal bleeding; and circulatory collapse.
Essential workup and diagnostic testing
Diagnosis relies on oil immersion light microscopy of thick and thin Giemsa-stained blood smears, which demonstrate intraerythrocytic parasites. At least three negative smears over 48 hours are required to exclude malaria. Laboratory findings commonly include anemia, thrombocytopenia, leukocytopenia, elevated bilirubin and lactate dehydrogenase, electrolyte abnormalities, and evidence of renal or hepatic dysfunction. Chest radiography may reveal pulmonary edema. Advanced testing such as antigen detection assays or PCR can identify Plasmodium species and mixed infections. Lumbar puncture may be necessary to distinguish cerebral malaria from meningitis.
Differential diagnosis
The differential includes meningitis, encephalitis, sepsis, acute renal failure, viral hepatitis, acute hemolytic anemia, hypoglycemic coma, and heat stroke. In travelers or immigrants from endemic areas, malaria must be considered in any febrile illness.
Treatment and emergency management
Initial stabilization includes airway, breathing, and circulation management, isotonic fluid resuscitation for hypotension, active cooling for hyperthermia, and treatment of hypoglycemia or seizures as indicated. Antimalarial therapy depends on species, severity, and geographic resistance patterns, with artemisinin-based combination therapies recommended as first-line treatment worldwide. Severe P. falciparum infection requires intravenous artesunate. Supportive care for complications such as anemia, renal failure, and cerebral edema is critical.
Disposition and follow-up
ICU admission is indicated for severe P. falciparum malaria, high parasitemia, neurologic involvement, or inability to tolerate oral therapy. Patients with non–P. falciparum malaria who are clinically stable and able to take oral medications may be treated as outpatients with close follow-up.
Pearls and pitfalls
Malaria should always be considered in patients with fever and compatible exposure history, even months after travel. Fever patterns are unreliable, and early recognition is essential, as delays in treatment—especially with P. falciparum—are associated with high mortality.