- Published on
KembaraXtra-Medicine- Acute Coronary Syndrome
Acute coronary syndrome (ACS) refers to a spectrum of life-threatening clinical conditions caused by a sudden reduction in blood flow to the myocardium. It most commonly results from rupture or erosion of a vulnerable atherosclerotic plaque in a coronary artery, followed by platelet activation and thrombus formation. ACS carries high morbidity and mortality, often requiring immediate diagnosis and intervention. Modern management relies on standardized, protocol-driven medical and interventional strategies aimed at rapid restoration of coronary blood flow and prevention of recurrent ischemic events.
ACS encompasses three related clinical entities: ST-elevation myocardial infarction (STEMI), non–ST-elevation myocardial infarction (NSTEMI), and unstable angina (UA). STEMI represents the most severe form and is characterized by ST-segment elevation on electrocardiography (ECG) in the appropriate clinical setting, typically reflecting complete coronary artery occlusion. NSTEMI and UA fall under the category of non–ST-elevation ACS (NSTE-ACS) and are distinguished by the absence of ST-segment elevation; NSTEMI is defined by elevated cardiac biomarkers, whereas UA lacks biomarker elevation. With the advent of high-sensitivity troponin assays, most patients previously diagnosed with UA are now reclassified as NSTEMI, reinforcing the concept that ACS exists along a continuous spectrum of ischemic injury.
In the United States, cardiovascular disease remains the leading cause of death, accounting for approximately 640,000 deaths annually. Each year, an estimated 600,000 new myocardial infarctions and 200,000 recurrent events occur. Approximately 70% of myocardial infarctions are NSTEMI. Although STEMI has higher short-term mortality, patients with NSTE-ACS tend to have worse long-term outcomes because they are generally older and have a higher burden of comorbidities such as diabetes, hypertension, and prior coronary artery disease. The median age at presentation is about 68 years, and men are affected more frequently than women, though cardiovascular disease remains the leading cause of death among women.
Major risk factors for ACS mirror those for atherosclerotic coronary artery disease and include hypertension, diabetes mellitus, dyslipidemia, tobacco use, obesity, and a family history of premature coronary disease. Female-specific risk factors, such as pregnancy-related disorders and early menopause, also contribute. These factors promote endothelial dysfunction, plaque formation, and progression to plaque instability and rupture.
Clinically, ACS most often presents with chest discomfort described as pressure or heaviness, typically substernal, and often radiating to the neck, jaw, shoulders, or back. Symptoms may be accompanied by diaphoresis, dyspnea, nausea, or fatigue. Certain populations—including women, elderly patients, individuals with diabetes, and postoperative patients—frequently present atypically, sometimes without chest pain. Unstable angina may manifest as rest angina, new-onset severe angina, or previously stable angina that has become more frequent, prolonged, or severe. Physical examination findings alone are insufficient for diagnosis but are important for assessing hemodynamic stability and identifying complications such as heart failure or cardiogenic shock.
The pathophysiologic hallmark of ACS is rupture of a vulnerable atherosclerotic plaque with a thin fibrous cap and lipid-rich core. Plaque rupture triggers platelet aggregation and thrombus formation, leading to partial or complete coronary artery occlusion. STEMI typically results from complete occlusion, whereas NSTEMI usually reflects subtotal obstruction or transient occlusion. Interestingly, angiographic studies show that many culprit lesions were only moderately stenotic prior to rupture.
Diagnosis of ACS relies on a combination of clinical assessment, ECG findings, and cardiac biomarkers. A 12-lead ECG should be obtained promptly in all patients with suspected ACS, as it guides immediate management. Cardiac troponin is the gold-standard biomarker for myocardial injury, and serial measurements help distinguish acute infarction from other causes of myocardial damage. High-sensitivity troponin assays allow earlier diagnosis or exclusion of myocardial infarction, often within a few hours of presentation. Additional testing, such as chest radiography, echocardiography, or coronary CT angiography, may aid in diagnosis or exclude alternative causes of chest pain.
Risk stratification is a key component of ACS management. Clinical risk scores such as TIMI, GRACE, and HEART integrate patient characteristics, ECG changes, biomarker levels, and clinical presentation to estimate short- and intermediate-term risk. These tools help guide decisions regarding invasive versus conservative strategies and the urgency of coronary angiography.
Treatment of ACS focuses on relieving ischemia, preventing thrombus propagation, and reducing future cardiovascular risk. STEMI is a true medical emergency and requires immediate reperfusion therapy. Primary percutaneous coronary intervention (PCI) is preferred and should be performed within 90 minutes of first medical contact when available. If timely PCI is not possible, fibrinolytic therapy should be administered within 12 hours of symptom onset in eligible patients, followed by transfer to a PCI-capable center. In NSTE-ACS, management is guided by risk stratification, with high-risk patients benefiting from an early invasive strategy and lower-risk patients managed initially with intensive medical therapy.
Medical management includes dual antiplatelet therapy with aspirin and a P2Y12 inhibitor, anticoagulation, beta-blockers, nitrates, statins, and angiotensin-converting enzyme inhibitors or angiotensin receptor blockers when indicated. Oxygen is reserved for patients with hypoxemia, and morphine may be used cautiously for refractory pain. Long-term therapy after ACS emphasizes secondary prevention through aggressive risk-factor modification, adherence to guideline-directed medical therapy, and participation in cardiac rehabilitation programs.
All patients with ACS should be managed in close collaboration with cardiology specialists, and selected patients may require referral for coronary artery bypass grafting. Early recognition, rapid risk stratification, and timely reperfusion remain the cornerstones of improving survival and long-term outcomes in acute coronary syndrome.
Acute coronary syndrome (ACS) refers to a spectrum of life-threatening clinical conditions caused by a sudden reduction in blood flow to the myocardium. It most commonly results from rupture or erosion of a vulnerable atherosclerotic plaque in a coronary artery, followed by platelet activation and thrombus formation. ACS carries high morbidity and mortality, often requiring immediate diagnosis and intervention. Modern management relies on standardized, protocol-driven medical and interventional strategies aimed at rapid restoration of coronary blood flow and prevention of recurrent ischemic events.
ACS encompasses three related clinical entities: ST-elevation myocardial infarction (STEMI), non–ST-elevation myocardial infarction (NSTEMI), and unstable angina (UA). STEMI represents the most severe form and is characterized by ST-segment elevation on electrocardiography (ECG) in the appropriate clinical setting, typically reflecting complete coronary artery occlusion. NSTEMI and UA fall under the category of non–ST-elevation ACS (NSTE-ACS) and are distinguished by the absence of ST-segment elevation; NSTEMI is defined by elevated cardiac biomarkers, whereas UA lacks biomarker elevation. With the advent of high-sensitivity troponin assays, most patients previously diagnosed with UA are now reclassified as NSTEMI, reinforcing the concept that ACS exists along a continuous spectrum of ischemic injury.
In the United States, cardiovascular disease remains the leading cause of death, accounting for approximately 640,000 deaths annually. Each year, an estimated 600,000 new myocardial infarctions and 200,000 recurrent events occur. Approximately 70% of myocardial infarctions are NSTEMI. Although STEMI has higher short-term mortality, patients with NSTE-ACS tend to have worse long-term outcomes because they are generally older and have a higher burden of comorbidities such as diabetes, hypertension, and prior coronary artery disease. The median age at presentation is about 68 years, and men are affected more frequently than women, though cardiovascular disease remains the leading cause of death among women.
Major risk factors for ACS mirror those for atherosclerotic coronary artery disease and include hypertension, diabetes mellitus, dyslipidemia, tobacco use, obesity, and a family history of premature coronary disease. Female-specific risk factors, such as pregnancy-related disorders and early menopause, also contribute. These factors promote endothelial dysfunction, plaque formation, and progression to plaque instability and rupture.
Clinically, ACS most often presents with chest discomfort described as pressure or heaviness, typically substernal, and often radiating to the neck, jaw, shoulders, or back. Symptoms may be accompanied by diaphoresis, dyspnea, nausea, or fatigue. Certain populations—including women, elderly patients, individuals with diabetes, and postoperative patients—frequently present atypically, sometimes without chest pain. Unstable angina may manifest as rest angina, new-onset severe angina, or previously stable angina that has become more frequent, prolonged, or severe. Physical examination findings alone are insufficient for diagnosis but are important for assessing hemodynamic stability and identifying complications such as heart failure or cardiogenic shock.
The pathophysiologic hallmark of ACS is rupture of a vulnerable atherosclerotic plaque with a thin fibrous cap and lipid-rich core. Plaque rupture triggers platelet aggregation and thrombus formation, leading to partial or complete coronary artery occlusion. STEMI typically results from complete occlusion, whereas NSTEMI usually reflects subtotal obstruction or transient occlusion. Interestingly, angiographic studies show that many culprit lesions were only moderately stenotic prior to rupture.
Diagnosis of ACS relies on a combination of clinical assessment, ECG findings, and cardiac biomarkers. A 12-lead ECG should be obtained promptly in all patients with suspected ACS, as it guides immediate management. Cardiac troponin is the gold-standard biomarker for myocardial injury, and serial measurements help distinguish acute infarction from other causes of myocardial damage. High-sensitivity troponin assays allow earlier diagnosis or exclusion of myocardial infarction, often within a few hours of presentation. Additional testing, such as chest radiography, echocardiography, or coronary CT angiography, may aid in diagnosis or exclude alternative causes of chest pain.
Risk stratification is a key component of ACS management. Clinical risk scores such as TIMI, GRACE, and HEART integrate patient characteristics, ECG changes, biomarker levels, and clinical presentation to estimate short- and intermediate-term risk. These tools help guide decisions regarding invasive versus conservative strategies and the urgency of coronary angiography.
Treatment of ACS focuses on relieving ischemia, preventing thrombus propagation, and reducing future cardiovascular risk. STEMI is a true medical emergency and requires immediate reperfusion therapy. Primary percutaneous coronary intervention (PCI) is preferred and should be performed within 90 minutes of first medical contact when available. If timely PCI is not possible, fibrinolytic therapy should be administered within 12 hours of symptom onset in eligible patients, followed by transfer to a PCI-capable center. In NSTE-ACS, management is guided by risk stratification, with high-risk patients benefiting from an early invasive strategy and lower-risk patients managed initially with intensive medical therapy.
Medical management includes dual antiplatelet therapy with aspirin and a P2Y12 inhibitor, anticoagulation, beta-blockers, nitrates, statins, and angiotensin-converting enzyme inhibitors or angiotensin receptor blockers when indicated. Oxygen is reserved for patients with hypoxemia, and morphine may be used cautiously for refractory pain. Long-term therapy after ACS emphasizes secondary prevention through aggressive risk-factor modification, adherence to guideline-directed medical therapy, and participation in cardiac rehabilitation programs.
All patients with ACS should be managed in close collaboration with cardiology specialists, and selected patients may require referral for coronary artery bypass grafting. Early recognition, rapid risk stratification, and timely reperfusion remain the cornerstones of improving survival and long-term outcomes in acute coronary syndrome.


- Published on
KembaraXtra- Medicine – Acute Stress Disorder
Acute stress disorder (ASD) is a trauma-related condition characterized by severe acute stress reactions that occur between 3 days and 1 month following exposure to a traumatic event. Trauma exposure may be direct, witnessed, learned about when it occurs to a close family member or friend, or experienced through repeated or extreme exposure to traumatic details. ASD represents an early posttraumatic response and is distinct from posttraumatic stress disorder (PTSD), which is diagnosed when symptoms persist beyond one month.
The prevalence of ASD varies depending on the type of traumatic exposure. Rates are generally below 20% following non–interpersonal traumas such as motor vehicle accidents, traumatic brain injury, or severe burns. Prevalence is estimated at approximately 14% following war-related trauma and rises substantially, ranging from 20% to 50%, following interpersonal trauma such as sexual assault or witnessing mass violence. ASD occurs more frequently in females than in males. It can be diagnosed across the lifespan, although symptom expression differs by age; for example, children may experience trauma-related dreams without direct trauma content and may not verbalize fear. Genetic vulnerability may contribute, with differential serotonin transporter functioning (e.g., 5-HTTLPR variants) potentially influencing trauma responses. Higher prevalence among women may reflect both increased exposure to interpersonal violence and sex-related neurobiological differences in stress reactivity.
Risk factors for ASD include greater perceived severity of the trauma, catastrophic interpretations of the event, exaggerated expectations of future harm, hopelessness, high negative affect, avoidant coping styles, prior trauma exposure, lack of social support, and the presence of preexisting psychiatric disorders such as anxiety or depression. These factors influence both symptom onset and severity following trauma.
Diagnosis of ASD is based on DSM-5 criteria. Individuals must have been exposed to actual or threatened death, serious injury, or sexual violation and must experience nine or more symptoms from five symptom clusters: intrusion, negative mood, dissociation, avoidance, and arousal. Intrusion symptoms include distressing memories, nightmares, flashbacks, and psychological or physiological distress when exposed to trauma reminders. Negative mood is reflected in an inability to experience positive emotions. Dissociative symptoms include altered sense of reality or inability to recall aspects of the trauma. Avoidance involves efforts to evade trauma-related thoughts, feelings, or external reminders. Arousal symptoms include sleep disturbance, irritability, hypervigilance, difficulty concentrating, and exaggerated startle response. Symptoms must persist for at least 3 days and no longer than 1 month after trauma exposure and must cause clinically significant impairment in functioning. Symptoms must not be attributable to substances, medical conditions, or other psychiatric disorders such as brief psychotic disorder. If symptoms persist beyond one month, PTSD should be considered.
The etiology of ASD is multifactorial and not fully established. Dissociative models suggest that individuals cope with overwhelming trauma by restricting awareness of emotional experiences. Cognitive models emphasize maladaptive appraisals, avoidance, distraction, and dysfunctional beliefs about responsibility or threat. Biological theories focus on acute trauma-related alterations in neurobiological systems, including dysregulation of cortisol, catecholamines, glucocorticoids, serotonin, and endogenous opioids, which collectively mediate stress responses.
Diagnosis is made through clinical interviews that assess trauma exposure, symptom onset, duration, and functional impairment. Structured clinical interviews and standardized self-report measures may assist diagnosis and severity assessment. Common tools include the Acute Stress Disorder Scale, Acute Stress Disorder Interview, and Stanford Acute Stress Reaction Questionnaire. Because dissociation may interfere with memory of the trauma or emotional responses, collateral information from family members, medical providers, or therapists is often helpful. No laboratory tests or imaging studies are indicated for ASD.
Treatment emphasizes early, supportive, and trauma-informed care. Initial interventions focus on establishing a therapeutic alliance, normalizing emotional reactions, and addressing immediate distress. Resilience-oriented psychosocial interventions aim to improve tolerance of trauma-related distress, reduce avoidance behaviors, identify and manage triggers, improve sleep disturbances and nightmares, strengthen social support, reduce functional impairment, limit overgeneralization of danger, and correct maladaptive trauma-related beliefs. Early behavioral and educational interventions show small to moderate benefits in reducing trauma-related symptoms. In cases involving interpersonal violence, safety planning is essential when individuals remain in unsafe environments.
Psychotherapy is the first-line treatment for ASD. Evidence-based nonpharmacologic interventions include cognitive behavioral therapy (CBT), cognitive processing therapy (CPT), and relaxation-based approaches such as mindfulness. Pharmacologic treatment is generally reserved for individuals who do not respond adequately to psychotherapy. There are currently no FDA-approved medications for ASD, although clinicians may consider medications approved for PTSD, including selective serotonin reuptake inhibitors such as sertraline or paroxetine. Acute pharmacologic management may be necessary in emergencies involving severe agitation, dangerous behavior, or psychosis, using short-acting benzodiazepines or neuroleptics when appropriate. Antiadrenergic agents, including β-blockers, may help reduce arousal symptoms. Prazosin is recommended for trauma-related nightmares. Benzodiazepines may provide short-term relief for insomnia or anxiety, but prolonged use is discouraged due to associations with increased long-term PTSD risk.
Complementary approaches such as yoga, meditation, relaxation response breathing, progressive muscle relaxation, and mindfulness practices may be beneficial for symptom relief. Eye movement desensitization and reprocessing (EMDR) has shown promise in reducing ASD symptoms, although further research is needed to clarify its effects relative to natural recovery.
Most individuals with ASD can be safely managed in an outpatient setting. Higher levels of care, including partial or inpatient hospitalization, may be necessary for patients with severe symptoms, comorbid psychiatric or medical conditions, or suicidal or homicidal ideation. Care should be delivered in the least restrictive environment that ensures safety. Referral to a qualified mental health clinician is recommended for diagnosis and treatment.
Although ASD may precede PTSD in some individuals, many people with ASD recover without developing PTSD. Current evidence suggests that ASD criteria alone are not sufficient for predicting PTSD risk. Research indicates that focusing on symptom subtypes, particularly high arousal symptoms, may improve prediction of later PTSD. Culturally sensitive assessment tools and culturally humble clinical practices are essential when working with diverse populations. Trauma patients discharged within 72 hours of hospital admission should be rescreened after discharge to detect emerging symptoms.
Preventive interventions such as single-session psychological debriefing or critical incident stress debriefing have not demonstrated effectiveness and are no longer routinely recommended. In contrast, psychological first aid (PFA) focuses on promoting safety, calmness, social connection, and hope, and has demonstrated benefits in reducing negative affect and enhancing positive affect shortly after trauma.
- Published on
KembaraXtra-Medicine- Acute Colonic Pseudo-Obstruction (Ogilvie Syndrome)
Acute colonic pseudo-obstruction, also known as Ogilvie syndrome, is a condition characterized by acute, massive dilation of the cecum and right colon, with possible extension to the rectum, occurring in the absence of a mechanical obstruction. It is most commonly seen in hospitalized patients, particularly following surgery or in those with severe underlying medical illnesses. Cecal dilation greater than 12 cm on abdominal imaging is clinically significant, as it markedly increases the risk of spontaneous colonic ischemia and perforation. Certain medications, especially opioids and anticholinergics, also play an important role in precipitating this condition.
The true incidence and prevalence of Ogilvie syndrome are unknown. Available data suggest a predominance in older adults, with a higher frequency in men, typically presenting in the sixth decade of life. However, women may be affected at a younger age, particularly in the setting of obstetric complications such as cesarean delivery. Overall, risk increases with advancing age and the presence of serious comorbidities.
Multiple risk factors are associated with acute colonic pseudo-obstruction. Elderly patients and those with critical illness are at greatest risk. Common predisposing factors include recent surgery (especially orthopedic, spinal, or cardiac procedures), trauma, sepsis, electrolyte disturbances, metabolic derangements, neurologic disease, cardiac events, and immobility. Numerous medications—including opioids, anticholinergics, antipsychotics, antidepressants, calcium channel blockers, antiparkinsonian agents, and chemotherapeutic drugs—are strongly implicated. Obstetric conditions and infections such as cytomegalovirus and tuberculosis have also been associated.
Clinically, patients most often present with acute abdominal distention and crampy abdominal pain. The distention can be severe enough to impair respiration. Associated symptoms may include nausea, vomiting, constipation, obstipation, or paradoxical diarrhea, although bowel habits are variable. Notably, up to half of patients may continue to pass flatus. On physical examination, the abdomen is markedly distended and tympanic to percussion, with variable tenderness. Bowel sounds may be hypoactive or hyperactive. Peritoneal signs are typically absent early but are worrisome when present, as they may indicate ischemia or impending perforation.
The pathophysiology of Ogilvie syndrome is not fully understood, but it is believed to involve autonomic nervous system dysfunction. The ascending colon normally receives parasympathetic stimulation via the vagus nerve to promote motility, while sympathetic input inhibits motility. An imbalance—particularly reduced parasympathetic activity—appears to result in colonic atony and progressive dilation, although the precise mechanisms remain unclear.
The diagnosis of acute colonic pseudo-obstruction is primarily one of exclusion. Conditions that must be ruled out include mechanical bowel obstruction, volvulus, intussusception, ileus, and toxic megacolon. A careful history should focus on the progression of abdominal distention, timing of the last bowel movement or passage of flatus, recent surgeries, acute illnesses, and medication changes. Serial physical examinations are essential to monitor for evolving peritoneal signs.
Laboratory evaluation typically reveals electrolyte abnormalities such as hypokalemia, hypomagnesemia, or hypocalcemia, all of which can worsen colonic dysmotility and should be corrected. Leukocytosis and lactic acidosis may be present and are concerning for ischemia or impending perforation. Imaging plays a central role in diagnosis and monitoring. Plain abdominal radiographs are useful for assessing and tracking colonic dilation, while computed tomography or contrast-enhanced studies are essential to exclude mechanical obstruction and confirm the diagnosis.
The primary goals of treatment are decompression of the colon, relief of symptoms, and prevention of ischemia or perforation. Initial management is supportive in stable patients without severe pain or cecal dilation greater than 12 cm. This includes bowel rest, intravenous fluids, correction of electrolyte abnormalities, discontinuation of offending medications, nasogastric and rectal tube decompression, frequent repositioning, encouragement of ambulation when possible, and close monitoring with serial examinations and imaging.
If conservative measures fail or colonic dilation progresses, pharmacologic therapy with intravenous neostigmine is recommended in appropriate candidates. Neostigmine is an acetylcholinesterase inhibitor that enhances parasympathetic activity and induces rapid colonic decompression in most patients. Because of the risk of bradycardia and bronchospasm, administration requires close cardiovascular monitoring, with atropine readily available. Alternative agents such as methylnaltrexone may be considered in opioid-related cases, although supporting evidence is limited.
Colonoscopic decompression is another effective option when supportive care and pharmacologic therapy are unsuccessful or contraindicated. This approach has a high success rate but carries a small risk of perforation and mortality. Placement of a decompression tube during colonoscopy may reduce recurrence, although definitive evidence is lacking. Minimally invasive alternatives such as percutaneous cecostomy are reserved for refractory cases.
Surgical intervention is considered a last resort and is indicated in patients with perforation, ischemia, peritonitis, or failure of less invasive therapies. Surgical options range from cecostomy to segmental colectomy, depending on the severity of disease and presence of complications.
Patients with Ogilvie syndrome require close inpatient monitoring until bowel function returns and abdominal distention resolves. Management is best accomplished through a multidisciplinary approach involving gastroenterology and surgery. Preventive strategies in high-risk and critically ill patients include early mobilization, minimizing opioid and vasoactive drug use, correcting metabolic abnormalities, promoting early enteral feeding, and encouraging timely bowel movements.
Early recognition and prompt, stepwise management are essential, as untreated acute colonic pseudo-obstruction can progress rapidly to ischemia, perforation, and death, whereas appropriate therapy is often highly effective.
Acute colonic pseudo-obstruction, also known as Ogilvie syndrome, is a condition characterized by acute, massive dilation of the cecum and right colon, with possible extension to the rectum, occurring in the absence of a mechanical obstruction. It is most commonly seen in hospitalized patients, particularly following surgery or in those with severe underlying medical illnesses. Cecal dilation greater than 12 cm on abdominal imaging is clinically significant, as it markedly increases the risk of spontaneous colonic ischemia and perforation. Certain medications, especially opioids and anticholinergics, also play an important role in precipitating this condition.
The true incidence and prevalence of Ogilvie syndrome are unknown. Available data suggest a predominance in older adults, with a higher frequency in men, typically presenting in the sixth decade of life. However, women may be affected at a younger age, particularly in the setting of obstetric complications such as cesarean delivery. Overall, risk increases with advancing age and the presence of serious comorbidities.
Multiple risk factors are associated with acute colonic pseudo-obstruction. Elderly patients and those with critical illness are at greatest risk. Common predisposing factors include recent surgery (especially orthopedic, spinal, or cardiac procedures), trauma, sepsis, electrolyte disturbances, metabolic derangements, neurologic disease, cardiac events, and immobility. Numerous medications—including opioids, anticholinergics, antipsychotics, antidepressants, calcium channel blockers, antiparkinsonian agents, and chemotherapeutic drugs—are strongly implicated. Obstetric conditions and infections such as cytomegalovirus and tuberculosis have also been associated.
Clinically, patients most often present with acute abdominal distention and crampy abdominal pain. The distention can be severe enough to impair respiration. Associated symptoms may include nausea, vomiting, constipation, obstipation, or paradoxical diarrhea, although bowel habits are variable. Notably, up to half of patients may continue to pass flatus. On physical examination, the abdomen is markedly distended and tympanic to percussion, with variable tenderness. Bowel sounds may be hypoactive or hyperactive. Peritoneal signs are typically absent early but are worrisome when present, as they may indicate ischemia or impending perforation.
The pathophysiology of Ogilvie syndrome is not fully understood, but it is believed to involve autonomic nervous system dysfunction. The ascending colon normally receives parasympathetic stimulation via the vagus nerve to promote motility, while sympathetic input inhibits motility. An imbalance—particularly reduced parasympathetic activity—appears to result in colonic atony and progressive dilation, although the precise mechanisms remain unclear.
The diagnosis of acute colonic pseudo-obstruction is primarily one of exclusion. Conditions that must be ruled out include mechanical bowel obstruction, volvulus, intussusception, ileus, and toxic megacolon. A careful history should focus on the progression of abdominal distention, timing of the last bowel movement or passage of flatus, recent surgeries, acute illnesses, and medication changes. Serial physical examinations are essential to monitor for evolving peritoneal signs.
Laboratory evaluation typically reveals electrolyte abnormalities such as hypokalemia, hypomagnesemia, or hypocalcemia, all of which can worsen colonic dysmotility and should be corrected. Leukocytosis and lactic acidosis may be present and are concerning for ischemia or impending perforation. Imaging plays a central role in diagnosis and monitoring. Plain abdominal radiographs are useful for assessing and tracking colonic dilation, while computed tomography or contrast-enhanced studies are essential to exclude mechanical obstruction and confirm the diagnosis.
The primary goals of treatment are decompression of the colon, relief of symptoms, and prevention of ischemia or perforation. Initial management is supportive in stable patients without severe pain or cecal dilation greater than 12 cm. This includes bowel rest, intravenous fluids, correction of electrolyte abnormalities, discontinuation of offending medications, nasogastric and rectal tube decompression, frequent repositioning, encouragement of ambulation when possible, and close monitoring with serial examinations and imaging.
If conservative measures fail or colonic dilation progresses, pharmacologic therapy with intravenous neostigmine is recommended in appropriate candidates. Neostigmine is an acetylcholinesterase inhibitor that enhances parasympathetic activity and induces rapid colonic decompression in most patients. Because of the risk of bradycardia and bronchospasm, administration requires close cardiovascular monitoring, with atropine readily available. Alternative agents such as methylnaltrexone may be considered in opioid-related cases, although supporting evidence is limited.
Colonoscopic decompression is another effective option when supportive care and pharmacologic therapy are unsuccessful or contraindicated. This approach has a high success rate but carries a small risk of perforation and mortality. Placement of a decompression tube during colonoscopy may reduce recurrence, although definitive evidence is lacking. Minimally invasive alternatives such as percutaneous cecostomy are reserved for refractory cases.
Surgical intervention is considered a last resort and is indicated in patients with perforation, ischemia, peritonitis, or failure of less invasive therapies. Surgical options range from cecostomy to segmental colectomy, depending on the severity of disease and presence of complications.
Patients with Ogilvie syndrome require close inpatient monitoring until bowel function returns and abdominal distention resolves. Management is best accomplished through a multidisciplinary approach involving gastroenterology and surgery. Preventive strategies in high-risk and critically ill patients include early mobilization, minimizing opioid and vasoactive drug use, correcting metabolic abnormalities, promoting early enteral feeding, and encouraging timely bowel movements.
Early recognition and prompt, stepwise management are essential, as untreated acute colonic pseudo-obstruction can progress rapidly to ischemia, perforation, and death, whereas appropriate therapy is often highly effective.


- Published on
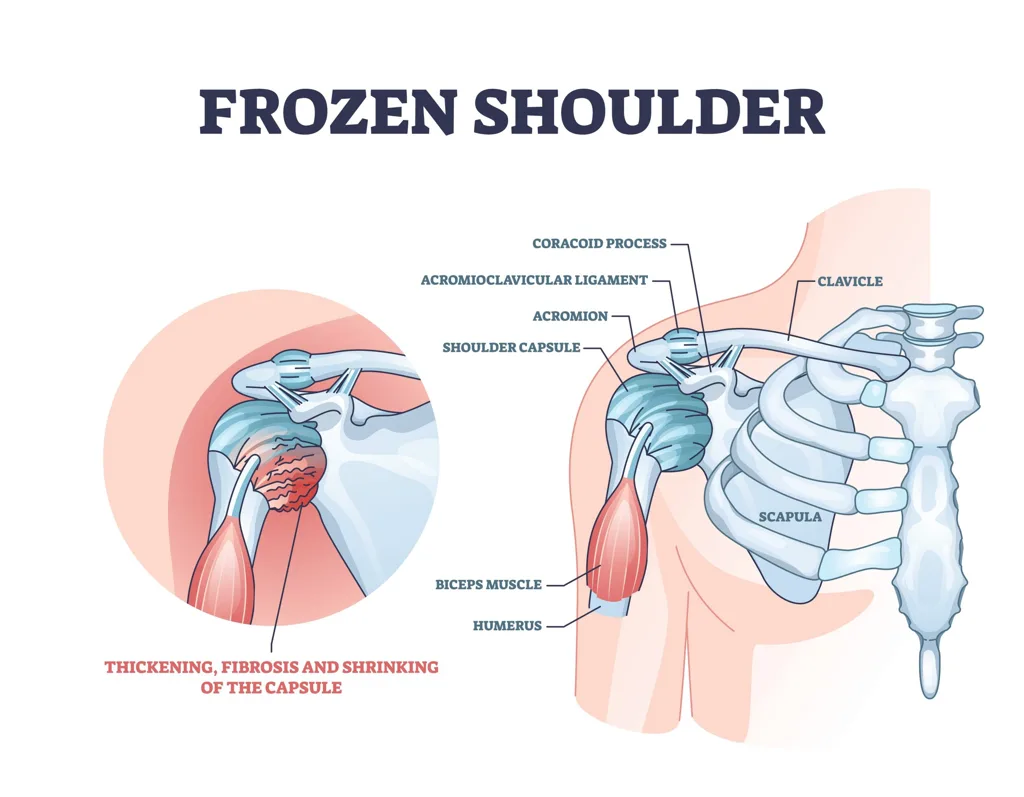
KembaraXtra- Medicine – Adhesive Capsulitis (Frozen Shoulder)
Adhesive capsulitis of the shoulder, commonly known as frozen shoulder, is a clinical diagnosis characterized by pain and progressive restriction of both active and passive range of motion of the glenohumeral joint. The condition is often idiopathic, but it has strong associations with systemic diseases, most notably diabetes mellitus. Other associated conditions include thyroid disorders, Dupuytren contracture, cervical disc disease, atherosclerosis, neurologic disorders such as stroke and Parkinson disease, and HIV infection treated with antiretroviral therapy. Secondary adhesive capsulitis may develop after shoulder trauma, most commonly proximal humerus fractures, or following shoulder surgery.
The incidence of adhesive capsulitis peaks in individuals in their mid-50s. The nondominant shoulder is more frequently affected, and 6% to 17% of patients develop symptoms in the contralateral shoulder within five years. The prevalence in the general population is approximately 2% to 5%, but it rises significantly to 10% to 20% among patients with diabetes. Females are affected more commonly than males.
Frozen shoulder typically follows a predictable natural history consisting of three phases. The initial painful or “freezing” phase is marked by gradual onset of severe shoulder pain, often worse at night and when lying on the affected side. This phase can last from one week to as long as ten months and is associated with minimal loss of motion. The second “frozen” or intermediate phase is dominated by stiffness and significant restriction in shoulder range of motion, particularly glenohumeral abduction and external rotation. Pain usually improves during this stage and is often present only at the extremes of motion or at night; this phase may last up to one year. The final “thawing” or recovery phase involves gradual reduction in pain and progressive improvement in range of motion over one to three years, although some patients experience persistent long-term limitations.
The underlying pathology of adhesive capsulitis is not completely understood. The most widely accepted theory involves a chronic inflammatory process leading to fibroblastic proliferation, resulting in thickening and contraction of the glenohumeral joint capsule, coracohumeral ligament, and rotator interval. This capsular fibrosis ultimately restricts joint motion and produces the characteristic stiffness of the condition.
Diagnosis is primarily based on history and physical examination, with the key distinguishing feature being restricted passive range of motion, which helps differentiate adhesive capsulitis from rotator cuff tears. The differential diagnosis includes rotator cuff disease, calcific tendinitis, glenohumeral osteoarthritis, septic arthritis, traumatic injuries such as dislocation or labral tears, and cervical spine or neurologic pathology. Imaging is not required for diagnosis but may be useful to exclude alternative causes. Plain radiographs are often obtained initially, while ultrasound or MRI may be used if clinical uncertainty persists. Characteristic MRI findings include thickening of the coracohumeral ligament, inferior joint capsule thickness greater than 5 mm, and loss of the axillary pouch.
Management is largely conservative, and patient education is critical. Patients should be counseled that frozen shoulder is generally self-limited but may take 18 to 24 months or longer to fully resolve. Initial treatment includes heat application and analgesics, most commonly nonsteroidal antiinflammatory drugs. Supervised physical therapy is a cornerstone of treatment and focuses on gentle, gradual passive stretching and capsular mobilization performed multiple times daily for at least three to six months. Aggressive strengthening exercises should be avoided in the early stages.
Intraarticular corticosteroid injections into the glenohumeral joint can provide significant short- to mid-term symptom relief, particularly when administered during the early phases of the disease. These injections often improve pain control and facilitate participation in physical therapy. Surgical intervention is rarely required and is generally reserved for patients who fail to improve after four to six months of consistent conservative management. Surgical options include manipulation under anesthesia and arthroscopic capsular release, with open release being rarely indicated.
The prognosis for adhesive capsulitis is generally favorable. Most patients ultimately regain approximately 90% of shoulder motion with significant pain improvement, although symptoms may persist for several months to more than three years. Recurrence in the same shoulder is uncommon, but involvement of the opposite shoulder occurs in a minority of patients, particularly those with diabetes. Referral to rheumatology or orthopedic surgery should be considered for patients who do not respond to appropriate conservative treatment.
Key clinical pearls include the importance of assessing both active and passive range of motion, recognizing that surgical treatment is rarely necessary, and understanding that early corticosteroid therapy combined with gentle physical therapy can significantly improve symptoms and patient satisfaction, especially in the early stages of the disease.
- Published on
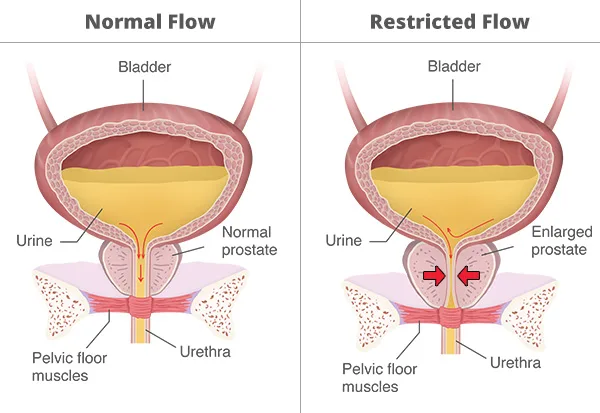
KembaraXtra- Medicine – Acute Urinary Retention
Acute urinary retention (AUR) is defined as the sudden inability to voluntarily pass urine despite a full bladder. It is often painful, although pain may be absent in older adults, patients with neurologic disorders, or those with chronic urinary retention. On physical examination, the bladder is typically distended, palpable, and percussible in the suprapubic region. AUR is distinct from chronic urinary retention, in which patients can void but retain a significant postvoid residual volume and usually do not experience pain.
AUR is one of the most common urologic emergencies, occurring predominantly in aging men, particularly those over 60 years of age, though it can affect individuals of any age and sex. Over a five-year period, approximately 10% of men older than 70 years and nearly one-third of men older than 80 years will experience an episode of AUR. The prevalence is estimated at 40 per 100,000 men and 3 per 100,000 women, with rates increasing as life expectancy rises. No specific genetic predisposition has been identified.
Clinically, patients most often present with acute inability to urinate, suprapubic pain or pressure, and a strong urge to void. Some patients pass only small amounts of urine. Low back pain may occur, and in severe cases, flank pain and costovertebral angle tenderness may develop if high bladder pressures are transmitted to the upper urinary tract. Many patients report worsening lower urinary tract symptoms (LUTS) in the days to weeks preceding AUR, including urgency, nocturia, hesitancy, intermittency, weak stream, stranguria, and incontinence. In patients with cognitive impairment or limited communication, AUR may manifest as agitation, restlessness, confusion, or delirium. Delayed presentation can result in acute kidney injury, electrolyte abnormalities, nausea, and lower extremity edema.
The etiology of AUR is often multifactorial, especially in older adults. Episodes may be spontaneous or precipitated by an identifiable trigger. Spontaneous AUR most commonly results from progressive bladder outlet obstruction, particularly due to benign prostatic hyperplasia (BPH), but may also arise from urethral strictures or pelvic masses. Precipitated AUR occurs when an acute event superimposes on underlying risk factors, such as surgery (especially spinal, orthopedic, or urologic), trauma, excessive alcohol intake, urinary tract infection, constipation, or neurologic insults including stroke or spinal cord injury. Medications are frequent precipitants and include antihistamines, sympathomimetics, anticholinergics, opioids, and anesthetic agents.
Pathophysiologically, urinary retention generally arises from one or more of three mechanisms. These include increased bladder outlet obstruction, as seen with BPH, urethral strictures, malignancy, constipation, or obstructing clots; impaired detrusor muscle innervation or contractility, associated with diabetic neuropathy, spinal cord disease, or prolonged outlet obstruction; and bladder overdistension, which can impair detrusor function following anesthesia or anticholinergic use. In women, causes may include pelvic organ prolapse, fibroid tumors, malignancy, urethral strictures, postpartum vulvar edema, labial fusion, or pelvic masses. Infections such as prostatitis, cystitis, urethritis, genital herpes, and herpes zoster may also precipitate AUR.
The diagnosis of AUR is usually clinical, based on history and physical examination. Workup focuses on identifying the underlying cause and associated complications. A detailed history should assess baseline voiding patterns, prior episodes of retention, recent surgeries or trauma, medication use, neurologic symptoms, and signs of infection. Physical examination includes abdominal assessment for bladder distension, rectal examination to evaluate prostate size, sphincter tone, and fecal impaction, and focused neurologic evaluation to exclude spinal cord or cauda equina pathology. Pelvic examination is essential in women to assess for pelvic organ prolapse or masses.
Laboratory testing typically includes serum electrolytes, blood urea nitrogen, and creatinine to assess renal function, along with urinalysis and urine culture obtained after catheterization. Prostate-specific antigen testing is not recommended in the acute setting, as levels may be falsely elevated following catheter placement. Imaging with bladder ultrasound or bedside bladder scanning is commonly used to confirm urinary retention, though accuracy may be limited by ascites, obesity, pelvic fluid collections, or surgical implants. Additional imaging such as CT, MRI, or renal ultrasound is reserved for suspected pelvic masses, spinal pathology, hydronephrosis, or renal impairment.
Management of AUR requires prompt bladder decompression, most commonly with indwelling urethral catheterization. Coude-tip catheters may facilitate placement in men with prostatic enlargement. Clean intermittent catheterization is an alternative for selected patients with adequate dexterity and cognition. If urethral catheterization is unsuccessful or contraindicated, suprapubic catheter placement is indicated. After decompression, patients should be monitored for postdecompression hematuria and postobstructive diuresis, which may cause significant fluid and electrolyte shifts. Medications that contribute to retention should be discontinued whenever possible.
Following acute management, a voiding trial is typically attempted five to seven days after catheter placement. Alpha-adrenergic blockers improve the likelihood of successful catheter removal in men with BPH and are commonly initiated unless contraindicated. Persistent inability to void after a trial warrants urologic consultation and further evaluation, which may include cystoscopy or urodynamic testing. Patients who fail repeated trials may require intermittent catheterization or long-term catheter management.
Most patients can be safely discharged with close follow-up if renal function is stable and postobstructive diuresis is absent. Hospital admission is indicated for patients with sepsis, complicated urinary tract infection, acute kidney injury, significant electrolyte disturbances, malignancy-related obstruction, hematuria, or suspected spinal cord compression. Urologic referral is required for difficult catheterizations, recurrent AUR, or postoperative cases, while gynecologic referral is necessary when pelvic pathology is identified in women.
Prevention strategies include treating constipation, maintaining mobility, avoiding medications known to precipitate urinary retention, and careful medication selection in patients with BPH. In men with large prostates, long-term use of 5-alpha reductase inhibitors may reduce the risk of future AUR episodes.
- Published on
KembaraXtra- Medicine – Acute Tubular Necrosis
Acute tubular necrosis (ATN) is a form of intrinsic acute kidney injury (AKI) characterized by damage and dysfunction of renal tubular epithelial cells. It is the most common cause of AKI in hospitalized patients. ATN most often results from ischemic injury, nephrotoxic exposure, sepsis, or a combination of these mechanisms. The condition represents a final common pathway of sustained renal hypoperfusion or direct tubular toxicity.
ATN is highly prevalent in critically ill patients. In intensive care settings, sepsis and hypotension account for approximately 50% of cases. More than half of affected patients require renal replacement therapy (RRT). Mortality remains high, particularly among patients requiring hemodialysis, with reported in-hospital mortality rates of 40% to 60%. Outcomes are influenced by patient-specific risk factors, including advanced age, diabetes mellitus, chronic kidney disease, hypertension, use of nonsteroidal antiinflammatory drugs or other nephrotoxins, hypovolemia, and impaired renal perfusion. Early recognition is critical to prevent ongoing tubular injury from repeated nephrotoxic insults.
Clinical presentation is often nonspecific, and physical examination findings may be minimal. Ischemic ATN should be suspected in patients with a history of hemorrhage, prolonged hypotension, recent surgery, or sepsis. Nephrotoxic ATN is more likely in the setting of exposure to iodinated radiocontrast media, nephrotoxic medications, rhabdomyolysis, hemolysis, or plasma cell dyscrasias such as multiple myeloma. Classically, ischemic ATN progresses through three phases, although this pattern is variable. The initiation phase occurs over hours to days and involves renal hypoperfusion with declining glomerular filtration rate (GFR), rising blood urea nitrogen and creatinine levels, and reduced urine output. The maintenance phase, lasting one to two weeks, is marked by established tubular injury, stabilized but low GFR, oliguria or anuria, and complications such as hyperkalemia, metabolic acidosis, uremia, and fluid overload. Patients are particularly vulnerable to infection during this phase, and approximately one-quarter of ATN-related deaths occur here. The recovery phase begins after two weeks or longer and is characterized by tubular regeneration, improving renal function, and often a period of marked polyuria due to impaired concentrating ability.
Pathologically, ATN is defined by vacuolization and loss of the proximal tubular brush border, disruption of the basement membrane, sloughing of tubular epithelial cells, and obstruction of tubules by granular casts. Interstitial edema and mild leukocyte infiltration may also be present. These structural changes underlie the impaired reabsorption, secretion, and filtration seen clinically.
ATN typically develops 24 to 48 hours after the inciting insult, making careful review of hemodynamic events and medication exposure in the preceding days essential. Ischemic contributors include shock, prolonged prerenal azotemia, major surgery, and systemic hypotension, although ATN may also occur in normotensive states due to regional renal hypoperfusion. Nephrotoxic causes include a wide range of agents such as NSAIDs, antimicrobial drugs (including aminoglycosides, vancomycin, amphotericin B, and antivirals), chemotherapeutic agents, calcineurin inhibitors, ACE inhibitors, ARBs, intravenous immunoglobulin, iodinated contrast media, heavy metals, and illicit substances. Endogenous toxins from rhabdomyolysis or hemolysis and cast nephropathy in multiple myeloma are additional important causes.
The differential diagnosis of ATN includes prerenal azotemia, postrenal obstruction, renal vascular disorders, acute glomerulonephritis, acute interstitial nephritis, and atheroembolic renal disease. Laboratory evaluation typically reveals a urinalysis with low specific gravity, low urine osmolality, and the presence of muddy brown granular casts and renal tubular epithelial cells. Fractional excretion of sodium is usually greater than 1%, although exceptions occur, particularly in patients taking ACE inhibitors, ARBs, or NSAIDs, and in early sepsis or contrast-induced injury. Novel biomarkers for early detection of tubular injury are under investigation, including FDA-approved urinary markers that identify patients at risk for AKI, though their routine clinical use is still evolving.
Imaging studies are not diagnostic for ATN but are useful to exclude obstructive causes of AKI or identify contributing pathology. Renal ultrasound or bladder scanning is commonly performed for this purpose.
Management of ATN follows general principles of AKI care. Treatment is largely supportive, focusing on correction of hemodynamic abnormalities, optimization of intravascular volume, avoidance of further nephrotoxic exposure, and careful electrolyte and fluid management. Renal replacement therapy is initiated when indicated for refractory metabolic disturbances or volume overload, but studies have shown no clear benefit to early initiation of dialysis in the absence of standard indications. Diuretics do not improve survival or renal recovery but may be used to assist with fluid balance.
Renal recovery typically occurs within two to three weeks, though outcomes vary widely. Younger patients and those with nonoliguric ATN tend to have better prognoses. Some patients may require dialysis for several months before sufficient tubular regeneration occurs. Among survivors, approximately 5% remain permanently dialysis-dependent. Nephrology consultation is recommended for diagnosis, management, and decisions regarding renal replacement therapy.
Prevention of ATN centers on aggressive volume resuscitation in hypovolemic patients, avoidance or minimization of nephrotoxic agents, and careful use of iodinated contrast media with appropriate pre- and post-procedural isotonic fluid administration. Early identification of at-risk patients remains the most effective strategy for reducing the morbidity and mortality associated with acute tubular necrosis.

- Published on
KembaraXtra- Medicine – Acute Respiratory Failure (ARF)
Acute respiratory failure (ARF) is a clinical condition in which the respiratory system fails to maintain adequate gas exchange, resulting in impaired oxygenation, carbon dioxide elimination, or both. It is broadly classified into type I (hypoxemic) and type II (hypercapnic) respiratory failure, with additional recognized forms including type III, which is associated with atelectasis, and type IV, which results from hypoperfusion of respiratory muscles in patients with shock. Hypoxemic respiratory failure is defined by a low arterial partial pressure of oxygen, typically less than 60 mm Hg while breathing room air, whereas hypercapnic respiratory failure is characterized by an elevated arterial partial pressure of carbon dioxide greater than 45 mm Hg. These two abnormalities may coexist depending on the underlying disease mechanism. ARF may present acutely within minutes to hours or evolve more gradually over days in subacute or chronic forms. Hypoxemia is particularly dangerous because it can rapidly lead to tissue hypoxia, although tissue oxygen delivery also depends on hemoglobin concentration, cardiac output, and regional blood flow, not solely on arterial oxygen tension.
Acute respiratory failure is common and represents a major burden on health care systems. An estimated 1.9 million patients discharged from acute care hospitals meet diagnostic criteria for ARF. The incidence peaks during winter months, reflecting the increased prevalence of respiratory infections. Mortality risk is significantly higher in older adults and in patients with chronic comorbid diseases involving the cardiac, pulmonary, neurologic, renal, or hepatic systems.
From a pathophysiologic standpoint, type I (hypoxemic) respiratory failure results from mechanisms such as ventilation–perfusion mismatch, intrapulmonary shunting, diffusion impairment, or reduced inspired oxygen concentration. Common clinical syndromes include acute respiratory distress syndrome, cardiogenic pulmonary edema, pneumonia, interstitial lung disease, pulmonary embolism, atelectasis, alveolar hemorrhage, and carbon monoxide poisoning. Type II (hypercapnic) respiratory failure is primarily due to alveolar hypoventilation, which may result from airway obstruction, central nervous system depression, neuromuscular disease, chest wall restriction, or severe obesity. Type III respiratory failure is typically seen in postoperative or trauma patients and is related to lung collapse and regional hypoventilation, while type IV respiratory failure occurs in the setting of shock, where inadequate perfusion of respiratory muscles leads to ventilatory failure.
The clinical presentation of ARF reflects both systemic hypoxia and hypercapnia. Early findings may include dyspnea, tachypnea, tachycardia, anxiety, and use of accessory respiratory muscles. As respiratory failure progresses, patients may develop headache, confusion, somnolence, asterixis, and eventually coma. Severe hypoxemia can impair myocardial contractility and cerebral function within seconds, while advanced hypercapnia may lead to respiratory acidosis and depressed mental status.
Diagnosis of acute respiratory failure is made clinically and confirmed with arterial blood gas analysis. Arterial blood gases allow differentiation between hypoxemic and hypercapnic failure and help assess severity. The diagnostic workup should continue after initial stabilization and focus on identifying the underlying cause, as definitive therapy depends on treating the precipitating condition. Laboratory evaluation typically includes arterial blood gases, a complete blood count, and a basic metabolic panel. Imaging studies commonly include chest radiography and, when indicated, computed tomography of the chest with angiography to evaluate for pulmonary embolism. Chest ultrasound, ventilation–perfusion scanning, and other targeted studies may be used based on clinical suspicion.
Management of acute respiratory failure is initially supportive and prioritizes stabilization of oxygenation and ventilation. Hypoxemia is treated with supplemental oxygen, noninvasive ventilation, or invasive mechanical ventilation as needed. Hypercapnia often requires ventilatory support, and the choice between noninvasive ventilation and endotracheal intubation is guided by the severity of symptoms, degree of acid–base disturbance, mental status, and the presence of comorbid conditions. Noninvasive ventilation is contraindicated in patients with respiratory arrest, hemodynamic instability, or inability to protect the airway. Close monitoring is essential to detect early failure of noninvasive strategies and avoid delayed intubation.
Mechanical ventilation is indicated in patients with severe hypoxemia refractory to oxygen therapy, worsening hypercapnic respiratory acidosis, declining mental status, excessive work of breathing, or evidence of inadequate oxygen delivery such as rising lactate levels. High-flow nasal oxygen has emerged as an effective alternative in selected patients, improving comfort and potentially reducing the need for intubation. Compared with conventional oxygen therapy, high-flow nasal oxygen may also reduce reintubation rates when used after extubation.
In all cases, definitive management requires prompt identification and treatment of the underlying etiology, such as infection, heart failure, pulmonary embolism, airway obstruction, or neuromuscular disease. Referral to a pulmonologist is appropriate for patients with severe, complex, or refractory respiratory failure, as well as for guidance on advanced ventilatory strategies and long-term management.

- Published on
KembaraXtra- Medicine – Acute Respiratory Distress Syndrome (ARDS)
Acute respiratory distress syndrome (ARDS) is a form of noncardiogenic pulmonary edema caused by acute alveolar injury leading to diffuse lung inflammation, interstitial and alveolar edema, severe hypoxemia, and respiratory failure. The hallmark is refractory hypoxemia due to disruption of the alveolar-capillary barrier, resulting in protein-rich alveolar edema. Earlier diagnostic criteria (AECC 1994) required acute onset, PaO₂/FiO₂ ≤200, bilateral infiltrates on chest radiograph, and no evidence of congestive heart failure (PAWP ≤18 mm Hg or no clinical evidence of elevated left atrial pressure). The 2012 Berlin definition refined this and requires: onset within 1 week of an insult or new/worsening respiratory symptoms; bilateral opacities on chest imaging not fully explained by effusions, lobar/lung collapse, or nodules; respiratory failure not fully explained by cardiac failure/fluid overload (objective assessment such as echocardiography if no clear ARDS risk factor); and oxygenation severity categorized by PaO₂/FiO₂ with PEEP/CPAP ≥5 cm H₂O (with altitude correction if >1000 m). Severity is mild (200
- Published on
KembaraXtra- Medicine – Adult-Onset Still Disease
Adult-onset Still disease (AOSD) is a rare systemic inflammatory disorder characterized by episodes of high-spiking fever, an evanescent rash, and arthralgias/arthritis, often accompanied by lymphadenopathy, hepatosplenomegaly, and markedly elevated serum ferritin. Many patients report a prodromal sore throat, sometimes preceding systemic symptoms. The daily (quotidian) fever pattern is a striking feature, and inflammatory arthritis can become progressive and destructive over time, leading to joint damage and disability in a subset of patients.
AOSD has several synonyms, including Still disease, systemic juvenile idiopathic arthritis (systemic JIA), and Wissler (Wissler-Fanconi) syndrome. The ICD-10-CM code is M06.1. Because it is uncommon, definitive epidemiologic data are limited, but prevalence has been reported between 1 and 34 per 1,000,000, with incidence around 0.1 to 0.4 per 100,000. A bimodal age distribution is described, with peaks around 15–25 and 36–46 years, and there is no consistent gender predisposition.
Infectious triggers are suspected in genetically susceptible individuals, as onset or relapse may follow a viral-like prodrome with pharyngitis, malaise, fever, and rash, although no single organism has been consistently implicated. Studies suggest a genetic component, with some associations reported between AOSD and certain HLA types (including class II DRB115 and DRB112 for susceptibility, and class I B35 with self-limited disease, DR2/DR5 with more chronic disease), though findings have not been consistently verified across populations.
Clinically, the “cardinal” triad of AOSD is fever, arthralgias/arthritis, and rash, and it occurs in most patients. Fever typically follows a quotidian pattern with spikes often occurring in the late afternoon or evening, frequently exceeding 102°F (38.9°C) and lasting a few hours before returning to normal. A “double quotidian” pattern with a second early-morning spike occurs in some cases. The classic rash is a salmon-pink, nonpruritic, maculopapular eruption most often involving the trunk and proximal extremities. The rash may appear with fever and fade when afebrile, which can make it easy to miss; Koebner phenomenon can occur. Arthralgias and myalgias are very common, and inflammatory arthritis often presents as symmetric polyarthritis affecting larger joints, with up to half of patients developing progressive destructive arthritis. Other manifestations may include sore throat, lymphadenopathy, hepatosplenomegaly, and serositis. Cardiac involvement such as pericarditis can occur and, in severe cases, can progress to tamponade, warranting prompt recognition and treatment.
The pathogenesis is not fully defined, but the disease appears driven by innate immune dysregulation and abnormal macrophage activation. Elevated proinflammatory cytokines are commonly described, including IL-1, IL-6, IL-18, TNF-α, interferon-γ, and others. More recently, increased circulating markers related to neutrophil extracellular traps (NETs) have been observed in active disease, with reductions after treatment, though the clinical implications continue to be studied.
There is no single diagnostic test that is pathognomonic for AOSD. Diagnosis is clinical and laboratory-based and is fundamentally a diagnosis of exclusion, requiring evaluation for infectious, neoplastic, and other inflammatory causes of fever and systemic illness. Laboratory abnormalities often include neutrophilic leukocytosis (commonly WBC >12 with >80% PMNs), very elevated inflammatory markers (often ESR >50 and CRP markedly elevated), normocytic anemia, thrombocytosis, hypoalbuminemia, and mild transaminitis. Creatine kinase (CPK) is typically normal even when myalgias are prominent. Hyperferritinemia is a particularly suggestive feature, and ferritin levels can exceed five times the upper limit of normal. Autoantibodies typical of other rheumatologic diseases are usually absent, with negative rheumatoid factor and negative ANA supporting (but not confirming) the diagnosis.
Imaging can support assessment of systemic involvement and complications. Plain radiographs may show early nonspecific changes such as soft-tissue swelling, effusions, or periarticular osteopenia, while later disease can demonstrate destructive arthropathy—classically in the wrists with joint-space narrowing and eventual ankylosis, and in hips with progressive joint destruction. CT or MRI may demonstrate lymphadenopathy, hepatosplenomegaly, and early bony or soft-tissue findings. FDG PET/CT may show increased uptake in spleen, bone marrow, and reactive lymph nodes, though these findings are not specific and must be interpreted in context.
Multiple classification/diagnostic criteria sets exist. The Yamaguchi criteria are widely used because they are sensitive and practical. The Fautrel criteria are more specific but require measurement of glycosylated ferritin, which is not routinely available in many settings. Regardless of criteria, careful exclusion of mimics is essential.
Prognosis is often favorable overall, with reported 5-year survival around 90%–95%, but many patients require long-term treatment to control inflammation and prevent relapse or progressive joint damage. Clinical course typically falls into one of three patterns: a self-limited/monophasic course with remission within a year, an intermittent/relapsing course with recurrent flares separated by remission, or a chronic/progressive course dominated by persistent arthritis and erosive destruction. Features associated with progression to chronic articular disease include polyarthritis, large-joint involvement, and very high ferritin levels at presentation.
Treatment depends on severity and organ involvement. Mild disease may respond to full-dose NSAIDs alone, though sustained control occurs in a minority. Most patients require systemic glucocorticoids, particularly when systemic symptoms are prominent or there is significant organ involvement. For steroid-sparing and control of arthritis, methotrexate is commonly used. Biologic therapy is frequently effective, especially for refractory disease or patients who cannot taper steroids. IL-1 blockade (such as anakinra or canakinumab) is often highly effective for systemic symptoms and inflammation. IL-6 inhibitors (such as tocilizumab or sarilumab) and TNF inhibitors (such as etanercept or infliximab) are also used, sometimes in combination with methotrexate. Response is typically reassessed after 2–3 months, and nonresponders are often switched to a biologic with a different mechanism. After prolonged remission, cautious tapering may be attempted. In severe or refractory cases, other therapies (including IVIG, and less commonly agents like rituximab or abatacept) have been reported with limited evidence.
A critical complication to recognize is macrophage activation syndrome (MAS), also referred to as reactive hemophagocytic lymphohistiocytosis. It occurs in a notable minority of AOSD patients and can be life-threatening, with substantial mortality. MAS can resemble a severe AOSD flare, but clues that raise suspicion include cytopenias (leukopenia and thrombocytopenia), hypofibrinogenemia, hypertriglyceridemia, and sometimes a paradoxical normalization of ESR despite clinical worsening. Bone marrow aspiration demonstrating hemophagocytosis is considered the diagnostic gold standard. Initial management commonly includes pulse-dose corticosteroids, IVIG, and cytokine-directed biologic therapy (often IL-1 or IL-6 blockade), alongside intensive supportive care.


- Published on
KembaraXtra- Medicine – Alcohol Use Disorder
Alcohol use disorder (AUD) describes problematic patterns of alcohol use that lead to clinically significant impairment or distress, and it includes a spectrum from hazardous use to dependence and withdrawal syndromes. “Moderate drinking” is commonly defined as up to two standard drinks per day for men and one standard drink per day for women and adults older than 65 years. “Hazardous” or “at-risk” drinking is defined as more than 14 drinks per week or more than 4 drinks per occasion for men, and 4 or more drinks on one occasion or 8 or more drinks per week for women. Alcohol withdrawal is defined by the development of characteristic symptoms after cessation or reduction of heavy and prolonged alcohol use, including autonomic hyperactivity, tremor, insomnia, nausea/vomiting, transient hallucinations or illusions, psychomotor agitation, anxiety, and grand mal seizures; symptoms must cause clinically significant distress or impairment and not be better explained by another condition.
AUD is also referred to as alcohol dependence syndrome, alcoholism, substance abuse, and alcohol withdrawal syndrome. ICD-10-CM codes fall under F10 (mental and behavioral disorders due to alcohol use), including F10.1 (harmful use), F10.2 (dependence syndrome), F10.3 (withdrawal state), F10.4 (withdrawal state with delirium), F10.5 (psychotic disorder), and F10.6 (amnesic syndrome). In U.S. clinical settings, alcohol-related problems are suggested by the clinical history in roughly 15% to 20% of primary care and hospitalized patients, with substantial economic and health impacts. AUD prevalence in adults (≥18 years) is commonly cited around 7%, with peak incidence in early adulthood and a common initial treatment age range in the mid-30s to mid-40s. Lifetime risk is higher in men than women. Risk is increased by family history, and AUD is more common in certain ancestry groups as described in clinical epidemiology summaries.
Patients may present with a wide range of physical and clinical findings related to intoxication, chronic use, or complications. Common clinical clues include recurring minor trauma, gastrointestinal bleeding from gastritis or varices, pancreatitis (acute or chronic), liver disease, alcohol odor on breath, tremulousness, tachycardia, peripheral neuropathy, and recent memory loss. Etiology is multifactorial, with both genetic and social determinants contributing. Commonly described risk factors include unstable social environments, unemployment, divorce, recurrent depression, addiction to other substances (including tobacco), and occupational patterns such as working very long hours.
Diagnosis and initial evaluation rely heavily on screening and history. Screening tools commonly used include CAGE, AUDIT-C, TWEAK/TACE (especially in pregnancy), and CRAFFT (adolescents). CAGE asks about the need to cut down, annoyance by criticism, guilt, and eye-openers; a positive screen should prompt further assessment. AUDIT-C focuses on alcohol frequency and quantity and provides a structured score (0–12), with ≥4 in men and ≥3 in women suggesting problematic drinking requiring more evaluation. A single-question screen (“When was the last time you had more than X drinks in a day?” where X is 5 for men and 4 for women) can also identify unhealthy use when anchored to a recent timeframe. Laboratory tests alone do not diagnose AUD but are useful for identifying complications and nutritional deficiencies. Common findings include elevated gamma-glutamyltransferase (GGT), abnormal AST/ALT (which may be normal or low in advanced liver disease), low albumin, electrolyte abnormalities (hypophosphatemia, hypomagnesemia), macrocytosis with elevated MCV on CBC, positive stool occult blood when gastritis/varices are present, and low vitamin levels including folate, B12, B6, and especially thiamine (B1). Imaging is not routinely required unless trauma is suspected; abdominal ultrasound or CT may show fatty liver or cirrhosis in advanced disease.
Nonpharmacologic treatment is a foundation of care and includes structured psychosocial therapies such as twelve-step facilitation, cognitive behavioral therapy, and motivational enhancement therapy. Coexisting depression should be treated alongside alcohol cessation rather than delayed, because mood disorders commonly coexist and influence relapse risk. Detoxification is not sufficient as stand-alone care and should function as a bridge into a longer-term treatment plan.
Alcohol withdrawal syndrome is managed based on severity and timing from the last drink. Blood ethanol typically declines at about 20 mg/dL per hour. Early withdrawal (“tremulous state”) often begins within 6 to 8 hours after the last drink (or 12 to 48 hours after reduction), peaking around 24 to 36 hours, with tremor, insomnia, mild agitation, and tachycardia. Depending on support system, comorbidities, and risk factors for severe withdrawal, detoxification may occur in outpatient or inpatient settings. Inpatient care includes monitoring (often with standardized withdrawal charts), seizure precautions, and symptom-titrated sedation. Benzodiazepines are the cornerstone of withdrawal management and are used either as fixed-dose tapers or symptom-triggered regimens. Symptom-triggered protocols often use the CIWA-Ar scale to guide dosing; patients with mild symptoms can sometimes be monitored outpatient, while higher CIWA-Ar scores and high-risk histories warrant inpatient detoxification. Beta-blockers can help control tachycardia and hypertension but should not be used alone because they do not prevent seizures or delirium; clonidine can reduce autonomic symptoms in mild to moderate cases but similarly does not prevent seizures or delirium. Vitamin replacement is essential, especially thiamine, and thiamine should be administered before IV dextrose to reduce the risk of precipitating Wernicke encephalopathy. Hydration and correction of electrolytes (including magnesium and phosphate) are often required.
Alcoholic hallucinosis consists of predominantly auditory hallucinations (sometimes visual/tactile/olfactory) that often peak 24 to 36 hours after abstinence and can resemble a primary psychotic disorder, but typically occurs without the marked clouding of sensorium seen in delirium tremens. Withdrawal seizures usually occur within 7 to 30 hours after cessation (often peaking around 13 to 24 hours) and are typically generalized; focal deficits, prolonged confusion, or fever should prompt evaluation for alternative causes (including imaging and lumbar puncture when indicated). Delirium tremens (DTs) is the most severe withdrawal state, usually occurring within the first week after reduction or cessation of heavy intake, commonly peaking around 72 to 96 hours, and characterized by profound confusion, tremor, vivid hallucinations, and marked autonomic hyperactivity; untreated mortality is significant. DTs require close monitoring, aggressive supportive care, and adequate benzodiazepine sedation (sometimes with agents such as lorazepam, chlordiazepoxide, diazepam, or midazolam in titratable settings), along with correction of fluids, electrolytes, and coexisting medical problems.
Long-term (chronic) treatment focuses on relapse prevention and sustained recovery using both psychosocial support and, when appropriate, pharmacotherapy. Acamprosate supports maintenance of abstinence after detoxification and is typically dosed as 666 mg three times daily; it should be avoided in severe renal impairment. Naltrexone (oral or extended-release monthly injection) reduces the rewarding effects of alcohol and can reduce heavy drinking; it must be avoided in acute hepatitis/hepatic failure and in people using opioids, as it can precipitate opioid withdrawal. Disulfiram causes an aversive reaction to alcohol ingestion by blocking aldehyde dehydrogenase, but is now used less often. Agents such as topiramate, baclofen, or gabapentin may be considered second line in moderate to severe AUD, with some guidance suggesting topiramate may be preferable in certain cases. Pharmacologic therapy is generally avoided in pregnant or breastfeeding individuals.
Disposition and referral are key components of management. Community supports such as Alcoholics Anonymous, Adult Children of Alcoholics, and family supports like Al-Anon or Al-A-Teen can be helpful. Inpatient detoxification is relatively indicated for patients with a history of DTs or withdrawal seizures, severe withdrawal symptoms, significant psychiatric or medical comorbidities, pregnancy, multiple prior detoxifications, recent high levels of alcohol intake, or lack of a reliable support network. Even when detoxification is successful, long-term outcomes depend heavily on engagement in ongoing treatment and social support, because relapse is common without sustained intervention.

