- Published on
Infectious Disease and Microbiology – Neurological Symptoms and Signs with Fever
Overview and Definitions
Clouding of consciousness refers to mild inattentiveness and slightly reduced wakefulness. Obtundation describes slowed responses to external stimuli with decreased alertness and increased sleepiness. Coma is a state of unarousable unconsciousness with no purposeful response to pain. Encephalitis is inflammation of the brain associated with neurologic or mental status changes. Meningitis is inflammation of the meninges surrounding the central nervous system and may be acute, presenting over hours, or chronic, lasting longer than four weeks.
Clinical Approach
Fever with neurologic symptoms requires urgent evaluation for central nervous system infection, systemic infection, or noninfectious neurologic pathology. History should include onset and progression of neurologic symptoms, medication and substance use, trauma, travel, animal exposures, immunocompromised states, and past medical history, with collateral history when available. A complete physical examination should be performed with emphasis on dermatologic, head and neck, cardiopulmonary, and a comprehensive neurologic assessment including mental status, cranial nerves, funduscopy, gait, tone, power, reflexes, sensation, and cerebellar function. Level of consciousness should be documented using the Glasgow Coma Scale. Examination for meningitis includes assessment of neck stiffness, Kernig and Brudzinski signs, and jolt accentuation of headache. Funduscopic examination is important to detect papilledema, subarachnoid hemorrhage, or hypertensive encephalopathy.
Epidemiology
Herpes simplex virus type 1 accounts for approximately 10% of encephalitis cases. In regions where tuberculosis is endemic, tuberculomas are a frequent cause of intracranial mass lesions. The annual incidence of bacterial meningitis in the United States is approximately 1.5 per 100,000 population. In adults, the most common pathogens are Streptococcus pneumoniae, Neisseria meningitidis, and Listeria monocytogenes, with Staphylococcus aureus, including methicillin-resistant strains, increasingly recognized.
Etiology
Acute fever with neurologic signs suggests bacterial meningitis, encephalitis, or infectious intracranial lesions, all of which are medical emergencies. CNS granulomas may be caused by syphilis, cysticercosis, tuberculoma, fungal infections, or sarcoidosis. Enteroviruses are the most common cause of aseptic meningitis. Opportunistic infections are common in advanced HIV infection. Listeria rhombencephalitis presents with brainstem and cranial nerve involvement and is best identified on MRI. Parasitic causes include Echinococcus granulosus, schistosomiasis, amebiasis, and paragonimiasis. Fungal causes include Cryptococcus neoformans, Histoplasma, Coccidioides, Blastomyces, Aspergillus, mucormycosis, and Pseudallescheria. Viral cerebellar encephalitis may follow measles, varicella, Lyme disease, rabies, or legionellosis. Free-living amoebae such as Naegleria fowleri cause acute fulminant meningitis, while Acanthamoeba and Balamuthia cause chronic meningoencephalitis. Noninfectious causes include drug intoxication or withdrawal, malignancy, and subarachnoid hemorrhage.
Clinical Features
Bacterial meningitis typically presents with acute onset of fever, headache, neck stiffness, photophobia, nausea, and vomiting. Absence of headache and jolt accentuation makes bacterial meningitis unlikely, although individual clinical features lack sensitivity. Central nervous system tuberculosis is commonly associated with fever. Brucellosis may cause chronic granulomatous meningitis with constitutional symptoms and relevant animal exposure.
Diagnostic Evaluation
Initial investigations include complete blood count, electrolytes, renal and liver function tests, and blood cultures. Lumbar puncture is essential unless contraindicated by raised intracranial pressure or coagulopathy and should include opening pressure, cell count with differential, protein, glucose, Gram stain, culture, mycobacterial and fungal studies, cryptococcal antigen, syphilis testing, and PCR for herpes simplex or varicella zoster virus. Neuroimaging with CT or MRI is required to exclude space-occupying lesions, with MRI offering superior sensitivity for encephalitis and brainstem disease. In bacterial meningitis, cerebrospinal fluid typically shows elevated opening pressure, neutrophilic pleocytosis, high protein, and low or normal glucose. Viral meningitis usually demonstrates lower white cell counts with lymphocytic predominance and moderately elevated protein.
Management
Empiric treatment for suspected bacterial meningitis includes a third- or fourth-generation cephalosporin combined with vancomycin, with ampicillin added in patients over 50 years of age or immunocompromised to cover Listeria. Dexamethasone should be administered early when pneumococcal meningitis is suspected. Acyclovir should be initiated if herpes simplex encephalitis is a consideration. Antimicrobial therapy must not be delayed for imaging or lumbar puncture when meningitis is suspected.
Additional Care
Management often requires a multidisciplinary approach, with involvement of physiotherapy, occupational therapy, and speech and language pathology during recovery.
Follow-Up and Prognosis
Early recognition and prompt treatment are critical to reducing morbidity and mortality associated with infectious neurologic syndromes.
Complications
Complications include death, deafness, visual impairment, seizures, stroke, cognitive impairment, and increased intracranial pressure.
Overview and Definitions
Clouding of consciousness refers to mild inattentiveness and slightly reduced wakefulness. Obtundation describes slowed responses to external stimuli with decreased alertness and increased sleepiness. Coma is a state of unarousable unconsciousness with no purposeful response to pain. Encephalitis is inflammation of the brain associated with neurologic or mental status changes. Meningitis is inflammation of the meninges surrounding the central nervous system and may be acute, presenting over hours, or chronic, lasting longer than four weeks.
Clinical Approach
Fever with neurologic symptoms requires urgent evaluation for central nervous system infection, systemic infection, or noninfectious neurologic pathology. History should include onset and progression of neurologic symptoms, medication and substance use, trauma, travel, animal exposures, immunocompromised states, and past medical history, with collateral history when available. A complete physical examination should be performed with emphasis on dermatologic, head and neck, cardiopulmonary, and a comprehensive neurologic assessment including mental status, cranial nerves, funduscopy, gait, tone, power, reflexes, sensation, and cerebellar function. Level of consciousness should be documented using the Glasgow Coma Scale. Examination for meningitis includes assessment of neck stiffness, Kernig and Brudzinski signs, and jolt accentuation of headache. Funduscopic examination is important to detect papilledema, subarachnoid hemorrhage, or hypertensive encephalopathy.
Epidemiology
Herpes simplex virus type 1 accounts for approximately 10% of encephalitis cases. In regions where tuberculosis is endemic, tuberculomas are a frequent cause of intracranial mass lesions. The annual incidence of bacterial meningitis in the United States is approximately 1.5 per 100,000 population. In adults, the most common pathogens are Streptococcus pneumoniae, Neisseria meningitidis, and Listeria monocytogenes, with Staphylococcus aureus, including methicillin-resistant strains, increasingly recognized.
Etiology
Acute fever with neurologic signs suggests bacterial meningitis, encephalitis, or infectious intracranial lesions, all of which are medical emergencies. CNS granulomas may be caused by syphilis, cysticercosis, tuberculoma, fungal infections, or sarcoidosis. Enteroviruses are the most common cause of aseptic meningitis. Opportunistic infections are common in advanced HIV infection. Listeria rhombencephalitis presents with brainstem and cranial nerve involvement and is best identified on MRI. Parasitic causes include Echinococcus granulosus, schistosomiasis, amebiasis, and paragonimiasis. Fungal causes include Cryptococcus neoformans, Histoplasma, Coccidioides, Blastomyces, Aspergillus, mucormycosis, and Pseudallescheria. Viral cerebellar encephalitis may follow measles, varicella, Lyme disease, rabies, or legionellosis. Free-living amoebae such as Naegleria fowleri cause acute fulminant meningitis, while Acanthamoeba and Balamuthia cause chronic meningoencephalitis. Noninfectious causes include drug intoxication or withdrawal, malignancy, and subarachnoid hemorrhage.
Clinical Features
Bacterial meningitis typically presents with acute onset of fever, headache, neck stiffness, photophobia, nausea, and vomiting. Absence of headache and jolt accentuation makes bacterial meningitis unlikely, although individual clinical features lack sensitivity. Central nervous system tuberculosis is commonly associated with fever. Brucellosis may cause chronic granulomatous meningitis with constitutional symptoms and relevant animal exposure.
Diagnostic Evaluation
Initial investigations include complete blood count, electrolytes, renal and liver function tests, and blood cultures. Lumbar puncture is essential unless contraindicated by raised intracranial pressure or coagulopathy and should include opening pressure, cell count with differential, protein, glucose, Gram stain, culture, mycobacterial and fungal studies, cryptococcal antigen, syphilis testing, and PCR for herpes simplex or varicella zoster virus. Neuroimaging with CT or MRI is required to exclude space-occupying lesions, with MRI offering superior sensitivity for encephalitis and brainstem disease. In bacterial meningitis, cerebrospinal fluid typically shows elevated opening pressure, neutrophilic pleocytosis, high protein, and low or normal glucose. Viral meningitis usually demonstrates lower white cell counts with lymphocytic predominance and moderately elevated protein.
Management
Empiric treatment for suspected bacterial meningitis includes a third- or fourth-generation cephalosporin combined with vancomycin, with ampicillin added in patients over 50 years of age or immunocompromised to cover Listeria. Dexamethasone should be administered early when pneumococcal meningitis is suspected. Acyclovir should be initiated if herpes simplex encephalitis is a consideration. Antimicrobial therapy must not be delayed for imaging or lumbar puncture when meningitis is suspected.
Additional Care
Management often requires a multidisciplinary approach, with involvement of physiotherapy, occupational therapy, and speech and language pathology during recovery.
Follow-Up and Prognosis
Early recognition and prompt treatment are critical to reducing morbidity and mortality associated with infectious neurologic syndromes.
Complications
Complications include death, deafness, visual impairment, seizures, stroke, cognitive impairment, and increased intracranial pressure.
- Published on
Emergency and Acute Medicine – Lymphocytosis
Overview and Definition
Lymphocytosis is defined as an absolute lymphocyte count greater than 4,000 cells/mm³ in peripheral blood. It may be reactive or neoplastic in origin. Reactive lymphocytosis is caused by infectious or noninfectious conditions, whereas neoplastic lymphocytosis reflects underlying hematologic malignancy. Infectious causes include viral, bacterial, protozoal, and parasitic diseases, while noninfectious causes include drugs, autoimmune disorders, and certain endocrinopathies. Atypical lymphocytes are non-neoplastic, activated lymphocytes that appear in response to immunologic stimulation; they are typically large, with nucleoli, irregular cytoplasm, and may appear to “indent” surrounding red blood cells on smear.
Clinical Approach
History and physical examination are essential to distinguish reactive from neoplastic lymphocytosis. Age is an important clue: acute EBV infection is common in children and young adults, while chronic lymphocytic leukemia is more common after the fifth decade. Assess symptom duration, vaccination history, prior illnesses, and medication use. Evaluate risk factors for HIV and tuberculosis, including sexual exposure, intravenous drug use, close contacts, travel, homelessness, and institutionalization. A detailed travel history should assess risk for food-, water-, and arthropod-borne infections. Physical examination should focus on lymphadenopathy and organ involvement such as hepatomegaly or splenomegaly. Many clinically stable patients without systemic toxicity can be evaluated as outpatients. Peripheral blood smears should be reviewed by experienced personnel, as atypical lymphocytes may be subtle.
Epidemiology
Lymphocytosis is common across many clinical conditions. Evidence of prior EBV infection is present in over 90% of adults. Primary EBV infection is frequent and often asymptomatic in children. CMV seroprevalence increases with age, exceeding 90% in adults over 80 years. Acute HIV infection may present with atypical lymphocytosis and a mononucleosis-like illness. Tuberculosis remains highly prevalent worldwide, with a large reservoir of latent infection.
Etiology
Common infectious causes include EBV, CMV, HIV, adenovirus, influenza, hepatitis A and B, measles, mumps, rubella, dengue, rickettsial infections, pertussis, tuberculosis, Bartonella, Brucella, syphilis, Q fever, Mycoplasma pneumoniae, toxoplasmosis, malaria, and babesiosis. Noninfectious causes include drug and toxic reactions, post-perfusion syndrome, immunizations, radiation exposure, endocrine disorders such as thyrotoxicosis or adrenal insufficiency, autoimmune diseases, smoking, and stress-related catecholamine release. Malignant causes include acute and chronic lymphocytic leukemia, lymphomas, hairy cell leukemia, and paraneoplastic syndromes. Other causes include sarcoidosis, thymoma, myasthenia gravis, Guillain–Barré syndrome, serum sickness, graft rejection, and inherited immune disorders.
Clinical Features
Acute EBV, CMV, or HIV infection may present with fever, lymphadenopathy, pharyngitis, fatigue, and anorexia. EBV typically causes exudative pharyngitis and hepatosplenomegaly, while CMV more often presents with prolonged fever and less prominent organomegaly. Cat-scratch disease presents with localized lymphadenopathy near the inoculation site. Cycling (Pel–Ebstein) fevers raise concern for Hodgkin lymphoma. Generalized lymphadenopathy is seen in many infections, leukemias, lymphomas, and sarcoidosis. Massive splenomegaly suggests myeloproliferative disorders, lymphomas, malaria, visceral leishmaniasis, or storage diseases. Viral hepatitis may present with jaundice and right upper quadrant tenderness.
Diagnostic Evaluation
Peripheral blood smear review is essential, and prior blood counts should be reviewed to assess chronicity. EBV infection is characterized by atypical lymphocytosis, positive heterophile antibody testing, and mild transaminitis. The Monospot test is highly specific and moderately sensitive. EBV serology distinguishes acute from past infection. CMV diagnosis relies on IgM and IgG serology, with PCR reserved for immunocompromised patients. HIV testing includes antigen–antibody screening with confirmatory testing; PCR is required in suspected acute infection. Toxoplasmosis is diagnosed by serology. Respiratory viruses such as influenza are detected by nasopharyngeal swab, with PCR offering the highest sensitivity. If malignancy is suspected, further evaluation may include flow cytometry, bone marrow biopsy, and cytogenetic studies. Imaging is reserved for clinical indications such as suspected pneumonia, organomegaly, or focal neurologic findings.
Management
Treatment is directed at the underlying cause. Uncomplicated EBV infection requires supportive care only. Patients with hepatosplenomegaly should avoid contact sports for up to two months due to risk of splenic rupture. Antibiotics such as ampicillin may cause rash in EBV infection and should be avoided. CMV treatment is generally reserved for immunocompromised patients. HIV infection requires antiretroviral therapy under specialist care.
Medications
Supportive therapy with acetaminophen or NSAIDs is appropriate for uncomplicated viral infections. Corticosteroids may be used for airway compromise in EBV. Antiviral therapy for CMV includes ganciclovir or valganciclovir in selected patients. Herpesvirus infections may be treated with acyclovir or valacyclovir. Influenza is treated with neuraminidase inhibitors when indicated.
Follow-Up and Prognosis
Patients should be counseled regarding warning signs and complications, particularly in EBV infection. Liver enzymes should be monitored until normalization. Persistent or unexplained lymphocytosis warrants hematology consultation to exclude malignancy.
Overview and Definition
Lymphocytosis is defined as an absolute lymphocyte count greater than 4,000 cells/mm³ in peripheral blood. It may be reactive or neoplastic in origin. Reactive lymphocytosis is caused by infectious or noninfectious conditions, whereas neoplastic lymphocytosis reflects underlying hematologic malignancy. Infectious causes include viral, bacterial, protozoal, and parasitic diseases, while noninfectious causes include drugs, autoimmune disorders, and certain endocrinopathies. Atypical lymphocytes are non-neoplastic, activated lymphocytes that appear in response to immunologic stimulation; they are typically large, with nucleoli, irregular cytoplasm, and may appear to “indent” surrounding red blood cells on smear.
Clinical Approach
History and physical examination are essential to distinguish reactive from neoplastic lymphocytosis. Age is an important clue: acute EBV infection is common in children and young adults, while chronic lymphocytic leukemia is more common after the fifth decade. Assess symptom duration, vaccination history, prior illnesses, and medication use. Evaluate risk factors for HIV and tuberculosis, including sexual exposure, intravenous drug use, close contacts, travel, homelessness, and institutionalization. A detailed travel history should assess risk for food-, water-, and arthropod-borne infections. Physical examination should focus on lymphadenopathy and organ involvement such as hepatomegaly or splenomegaly. Many clinically stable patients without systemic toxicity can be evaluated as outpatients. Peripheral blood smears should be reviewed by experienced personnel, as atypical lymphocytes may be subtle.
Epidemiology
Lymphocytosis is common across many clinical conditions. Evidence of prior EBV infection is present in over 90% of adults. Primary EBV infection is frequent and often asymptomatic in children. CMV seroprevalence increases with age, exceeding 90% in adults over 80 years. Acute HIV infection may present with atypical lymphocytosis and a mononucleosis-like illness. Tuberculosis remains highly prevalent worldwide, with a large reservoir of latent infection.
Etiology
Common infectious causes include EBV, CMV, HIV, adenovirus, influenza, hepatitis A and B, measles, mumps, rubella, dengue, rickettsial infections, pertussis, tuberculosis, Bartonella, Brucella, syphilis, Q fever, Mycoplasma pneumoniae, toxoplasmosis, malaria, and babesiosis. Noninfectious causes include drug and toxic reactions, post-perfusion syndrome, immunizations, radiation exposure, endocrine disorders such as thyrotoxicosis or adrenal insufficiency, autoimmune diseases, smoking, and stress-related catecholamine release. Malignant causes include acute and chronic lymphocytic leukemia, lymphomas, hairy cell leukemia, and paraneoplastic syndromes. Other causes include sarcoidosis, thymoma, myasthenia gravis, Guillain–Barré syndrome, serum sickness, graft rejection, and inherited immune disorders.
Clinical Features
Acute EBV, CMV, or HIV infection may present with fever, lymphadenopathy, pharyngitis, fatigue, and anorexia. EBV typically causes exudative pharyngitis and hepatosplenomegaly, while CMV more often presents with prolonged fever and less prominent organomegaly. Cat-scratch disease presents with localized lymphadenopathy near the inoculation site. Cycling (Pel–Ebstein) fevers raise concern for Hodgkin lymphoma. Generalized lymphadenopathy is seen in many infections, leukemias, lymphomas, and sarcoidosis. Massive splenomegaly suggests myeloproliferative disorders, lymphomas, malaria, visceral leishmaniasis, or storage diseases. Viral hepatitis may present with jaundice and right upper quadrant tenderness.
Diagnostic Evaluation
Peripheral blood smear review is essential, and prior blood counts should be reviewed to assess chronicity. EBV infection is characterized by atypical lymphocytosis, positive heterophile antibody testing, and mild transaminitis. The Monospot test is highly specific and moderately sensitive. EBV serology distinguishes acute from past infection. CMV diagnosis relies on IgM and IgG serology, with PCR reserved for immunocompromised patients. HIV testing includes antigen–antibody screening with confirmatory testing; PCR is required in suspected acute infection. Toxoplasmosis is diagnosed by serology. Respiratory viruses such as influenza are detected by nasopharyngeal swab, with PCR offering the highest sensitivity. If malignancy is suspected, further evaluation may include flow cytometry, bone marrow biopsy, and cytogenetic studies. Imaging is reserved for clinical indications such as suspected pneumonia, organomegaly, or focal neurologic findings.
Management
Treatment is directed at the underlying cause. Uncomplicated EBV infection requires supportive care only. Patients with hepatosplenomegaly should avoid contact sports for up to two months due to risk of splenic rupture. Antibiotics such as ampicillin may cause rash in EBV infection and should be avoided. CMV treatment is generally reserved for immunocompromised patients. HIV infection requires antiretroviral therapy under specialist care.
Medications
Supportive therapy with acetaminophen or NSAIDs is appropriate for uncomplicated viral infections. Corticosteroids may be used for airway compromise in EBV. Antiviral therapy for CMV includes ganciclovir or valganciclovir in selected patients. Herpesvirus infections may be treated with acyclovir or valacyclovir. Influenza is treated with neuraminidase inhibitors when indicated.
Follow-Up and Prognosis
Patients should be counseled regarding warning signs and complications, particularly in EBV infection. Liver enzymes should be monitored until normalization. Persistent or unexplained lymphocytosis warrants hematology consultation to exclude malignancy.

- Published on
KembaraXtra-Medicine: Abusive Head Trauma
Abusive head trauma (AHT) refers to traumatic brain injury resulting from the deliberate application of force to an infant or young child, most commonly under 4 years of age. Formerly termed shaken baby syndrome, AHT may result from shaking, blunt impact, or a combination of both, and the injury patterns do not reliably distinguish between these mechanisms. AHT is a life-threatening condition, and survivors frequently experience severe, lifelong neurologic sequelae. The most characteristic injury pattern includes subdural hemorrhage, often accompanied by extensive retinal hemorrhages and encephalopathy, although children may present with or without additional cutaneous, musculoskeletal, or visceral injuries. Diagnosis is often challenging because symptoms may be vague, caregivers may provide no history or an inconsistent history of trauma, and external signs of injury may be absent.
AHT is also known as shaken baby syndrome, infant whiplash syndrome, inflicted pediatric neurotrauma, or nonaccidental head injury. Relevant ICD-10-CM codes include T74.4 (shaken infant syndrome), T74.12 (child physical abuse, confirmed), and T76.12 (child physical abuse, suspected).
Epidemiologically, the incidence of AHT is estimated at 32–38 cases per 100,000 children under 1 year of age. It is the most common cause of fatal head injury in children under 2 years, accounting for more than half of serious or fatal traumatic injuries in this age group, and it is the leading cause of death from physical abuse overall. Mortality approaches 25%, and among survivors, long-term neurologic disability occurs in approximately 70%, including visual impairment, cognitive deficits, seizures, cerebral palsy, and intellectual disability. Males are affected more frequently than females, with reported ratios as high as 2:1.
Victims of AHT are typically younger than 1 year, with a median age of about 4 months. The peak incidence of fatal AHT occurs even earlier, between 1 and 2 months of age, which coincides with the normal developmental peak in infant crying. Infants are particularly vulnerable due to their relatively large head size, weak neck musculature, neural immaturity, and unique neuroanatomy, all of which increase susceptibility to acceleration–deceleration and rotational forces.
Risk factors for AHT include prematurity, perinatal illness, multiple gestations, male sex, excessive crying, and a range of caregiver and social factors such as young parental age, low educational attainment, substance use, mental illness, intimate partner violence, family disruption, social isolation, low socioeconomic status, and lack of affordable child care. Perpetrators are most often male caregivers, frequently the child’s father or the mother’s partner, although AHT occurs across all socioeconomic groups and may be inflicted by caregivers of any gender.
Clinically, infants with AHT often present with nonspecific symptoms such as lethargy, irritability, vomiting, poor feeding, seizures, apnea, or abnormal breathing. Caregivers may report no trauma or describe a minor fall inconsistent with the severity of injury. Presentations range from subtle symptoms to coma or cardiopulmonary arrest. Physical examination may reveal bruising, intraoral injuries such as a torn frenulum, macrocephaly, or a bulging fontanelle, but many infants have no external signs of injury.
Subdural hemorrhage is the most common neuroimaging finding in AHT and is one of the leading causes of subdural bleeding in infancy. Additional findings may include cerebral edema, loss of gray–white matter differentiation, parenchymal injury, torn or thrombosed bridging veins, cervical spine ligamentous injury, and spinal subdural hematomas. Skeletal surveys may reveal posterior rib fractures or classic metaphyseal lesions, and dilated ophthalmologic examination often shows retinal hemorrhages that are bilateral, multilayered, too numerous to count, and extending to the ora serrata, though some affected children may have no retinal hemorrhages. Retinoschisis may also be present. AHT is missed on initial presentation in roughly 30% of cases, often leading to repeated and escalating injury before diagnosis. Bias contributes to missed diagnoses, particularly among children from intact or socioeconomically advantaged families, underscoring that diagnosis must be based on clinical findings, not demographics.
The pathophysiology of AHT is thought to involve rotational deceleration forces, such as those generated during shaking, which damage bridging veins and result in subdural hemorrhage. Blunt impact can produce similar injury patterns. Severe cases may involve traumatic axonal injury, and vitreoretinal traction explains the characteristic retinal hemorrhages and retinoschisis. Many victims experience recurrent episodes of abuse, highlighting the importance of early recognition to prevent further harm.
The differential diagnosis includes accidental head trauma, severe infection, bleeding disorders, metabolic disorders, birth trauma, and brief resolved unexplained events (BRUE). Evaluation requires a thorough history and physical examination, including head circumference measurement, along with head CT for acute symptoms, followed by MRI of the brain and spine, ideally obtained several days after presentation. A skeletal survey is required, with repeat imaging after 2–3 weeks to identify healing fractures. A dilated ophthalmologic examination is essential. Laboratory testing is used to assess for coagulopathies, metabolic disorders, abdominal injury, and bone health, and social work evaluation is mandatory.
Management focuses on treating the child’s acute injuries and ensuring future safety. Most infants require hospital admission, often to an intensive care unit, for neurologic monitoring and supportive care. Long-term management includes consistent primary care, developmental surveillance, social services involvement, parenting support, and specialty referrals as indicated, including child abuse pediatrics, ophthalmology, neurology, neurosurgery, rehabilitation medicine, and early intervention services.
All health care providers are mandatory reporters of suspected child abuse. Every case of suspected AHT must be reported to child protective services, and many involve law enforcement. Other children in the household should undergo medical evaluation, as abuse is frequently recurrent. The role of the medical team is to objectively diagnose injury, not to determine legal responsibility.
Prevention efforts focus on parent and caregiver education, particularly around normal infant crying and the dangers of shaking. Programs such as the Period of Purple Crying, along with similar initiatives, aim to reduce AHT through anticipatory guidance, though results have been mixed. Broader public health strategies that reduce family stress—such as access to food assistance, health care, paid family leave, and affordable child care—are also important components of AHT prevention.


- Published on
KembaraXtra-Medicine: Absence Seizures
Absence seizures are a form of generalized seizure characterized by brief episodes of impaired consciousness, typically presenting as sudden staring spells. These events usually last 20 to 30 seconds or less, begin and end abruptly, and are not followed by post-ictal confusion. Patients are usually unaware that a seizure has occurred and immediately resume their prior activity. The classic electroencephalographic (EEG) hallmark is a generalized 3-Hz spike-and-wave discharge, often provoked by hyperventilation.
Absence seizures are also referred to as childhood absence epilepsy or simply absence seizures and are classified under ICD-10-CM code G40.309 (generalized idiopathic epilepsy, not intractable, without status epilepticus). They represent up to 18% of pediatric epilepsy syndromes, with an incidence of approximately 1 to 10 cases per 100,000 population. The condition is more common in girls than boys. Onset typically occurs between 4 and 10 years of age, with a peak incidence around 6 to 7 years, and seizures often remit spontaneously by early adulthood.
On physical and neurologic examination, patients with absence seizures are usually entirely normal. During seizures, patients are briefly unresponsive and may exhibit subtle motor features such as eye blinking, lip smacking, or small hand movements (automatisms). Unlike focal seizures, absence seizures are not associated with an aura, post-ictal confusion, or prolonged recovery. They may occur many times per day and are commonly triggered by hyperventilation, such as during exercise or intentional over-breathing during EEG testing. Tonic-clonic seizures are not typical of classic childhood absence epilepsy; if present, alternative epilepsy syndromes should be considered.
The etiology of absence seizures is considered genetic, with dysfunction in thalamocortical circuits leading to abnormal synchronous neuronal firing. Because of this generalized mechanism, seizures involve both hemispheres simultaneously, which explains the abrupt onset, brief duration, and lack of post-ictal symptoms.
Diagnosis relies heavily on EEG, which is essential and often diagnostic. EEG performed with hyperventilation and photic stimulation typically provokes the characteristic generalized 3-Hz spike-and-wave pattern. Ambulatory EEG or video EEG monitoring may be used when diagnostic uncertainty exists, particularly to distinguish absence seizures from focal seizures with impaired awareness or nonepileptic staring spells. In patients with a classic clinical presentation and EEG findings, neuroimaging is often unnecessary; however, brain MRI should be performed if the EEG is atypical or if focal features are suspected. CT scans should generally be avoided in children due to radiation exposure and low diagnostic yield.
The differential diagnosis includes juvenile absence epilepsy, juvenile myoclonic epilepsy, focal seizures with impaired awareness (complex partial seizures), and nonepileptic staring episodes. Key distinguishing features include seizure duration (seconds in absence seizures vs minutes in focal seizures), lack of post-ictal confusion, frequent daily occurrence, and the characteristic EEG findings. Correct differentiation is critical, as some antiseizure medications used for focal epilepsy can worsen absence seizures.
Treatment outcomes are generally excellent. Ethosuximide is considered the first-line therapy based on the best available evidence, followed by valproic acid and lamotrigine as alternatives. Ethosuximide is particularly favored because of its effectiveness and relatively narrow side-effect profile for absence seizures. Valproic acid should be used cautiously, especially in girls and women of childbearing potential, due to teratogenic risks. Carbamazepine and phenytoin are contraindicated, as they may exacerbate absence seizures or provoke absence status epilepticus.
Children with recurrent absence seizures require chronic antiseizure therapy, but prognosis is favorable. Many children become seizure-free and can be considered for medication withdrawal after 1 to 2 years without seizures. Absence seizures commonly remit during adolescence, and epilepsy may be considered resolved after 10 years without seizures, including at least 5 years off medication.
Patients with absence seizures should be referred to a child neurologist, ideally one with expertise in epilepsy, for diagnosis confirmation and long-term management. Families should be educated about seizure recognition, medication adherence, and safety considerations. Driving restrictions apply to patients with ongoing seizures, and regulations vary by jurisdiction. Preventive strategies include avoiding sleep deprivation and alcohol, both of which may lower seizure threshold.


- Published on
KembaraXtra-Medicine: Abdominal Compartment Syndrome
Abdominal compartment syndrome (ACS) is defined as organ dysfunction caused by increased intraabdominal pressure, also referred to as intraabdominal hypertension. When pressure within the abdomen rises, blood flow to vital organs falls, which can rapidly progress to multisystem organ failure and death if not recognized and treated promptly. The ICD-10-CM code is M79.A3 (Nontraumatic compartment syndrome of abdomen).
The true incidence of ACS outside trauma populations is not well defined, but among trauma patients it has been reported in roughly 1% to 14%, depending on the patient population and type of injury. ACS is most commonly seen in critically ill patients, and critical illness itself is the largest overarching risk factor. A particularly important trigger is large-volume intravenous fluid resuscitation, because “third-spacing” and tissue edema can increase intraabdominal pressure. As a result, ACS is often associated with severe burns, major trauma, postoperative states, and sepsis. It is also linked to intraabdominal or retroperitoneal conditions that increase abdominal contents or reduce abdominal wall compliance, including bowel distention/obstruction or ileus, pancreatitis with retroperitoneal edema, massive ascites, hemoperitoneum (such as after ruptured abdominal aortic aneurysm), liver transplantation, and large intraabdominal or retroperitoneal hematomas.
Clinically, the most striking physical finding is typically marked abdominal distention. Patients often develop difficulty with ventilation and decreased urine output, which are common early clues to organ compromise. Additional findings may reflect poor perfusion and shock physiology, including hypotension, cool extremities, skin mottling, and altered mental status. Many patients have abdominal tenderness and signs of volume overload such as edema and elevated jugular venous pressure, and they may deteriorate quickly with acute respiratory decompensation.
ACS can impair nearly every organ system because elevated intraabdominal pressure disrupts both perfusion and mechanics. Rising abdominal pressure can increase intracranial pressure, potentially worsening cerebral perfusion. Cardiovascular compromise can occur through reduced venous return from inferior vena cava compression and reduced ventricular compliance, leading to higher central venous and pulmonary pressures and reduced cardiac output. Respiratory failure is common because elevated abdominal pressure pushes the diaphragm upward, reducing chest wall compliance and tidal volumes, which contributes to atelectasis, hypoxemia, hypercarbia, and pneumonia. Mechanically ventilated patients may require higher airway pressures, increasing the risk of barotrauma. Renal dysfunction occurs as renal vein compression and renal arterial vasoconstriction reduce renal blood flow, producing oliguria or anuria. Reduced mesenteric perfusion may cause bowel ischemia and lactic acidosis.
A definitive diagnosis requires direct measurement of intraabdominal pressure, most commonly estimated using bladder pressure measured through a urinary catheter. For accuracy, measurements are ideally obtained with the patient supine, at end-expiration, and without active abdominal muscle contractions. While many research definitions use a threshold of >20 mm Hg with associated organ dysfunction, ACS may occur at lower pressures in susceptible patients. Oliguria commonly appears around 15 mm Hg, and anuria may occur near 30 mm Hg. Other measurement approaches (such as intragastric or intracolonic pressure estimation) exist but are used less commonly. Laboratory studies are generally not diagnostic, although lactic acidosis can suggest bowel ischemia and signals a worse prognosis. Imaging does not establish ACS, but chest imaging may show diaphragmatic elevation or pulmonary complications, and abdominal CT may sometimes demonstrate findings such as vena cava compression or bowel ischemia-related injury—these should be considered supportive rather than diagnostic.
Treatment focuses on supportive care and rapid reduction of intraabdominal pressure, with surgical decompression as the only definitive therapy when conservative measures fail or when pressures and organ dysfunction are severe. Supportive management includes aggressive hemodynamic and ventilatory support, along with measures to improve abdominal wall compliance. Patients should be kept supine when possible, since head-of-bed elevation can raise intraabdominal pressure. Nasogastric and rectal decompression may be required when bowel distention is contributing. Adequate sedation and analgesia help reduce abdominal wall muscle tone, and some patients need chemical paralysis to maximize relaxation. If tense ascites is a major contributor, large-volume paracentesis can substantially lower pressures. In patients with severe abdominal burns, escharotomy may be necessary to restore abdominal wall compliance. Mechanical ventilation is often challenging and may require low tidal volumes, permissive hypercapnia, high PEEP, and paralytics to maintain adequate gas exchange.
Although vasopressors, fluids, sedatives, analgesics, and paralytics are used to stabilize the patient, there are no medications that directly treat ACS, and diuretics do not play a therapeutic role even when patients appear volume overloaded. If intraabdominal pressure remains high with ongoing organ dysfunction, surgical decompression is performed, typically by opening the abdomen via a midline incision through the linea alba. In emergent situations this can be done at the bedside, followed by temporary abdominal closure (often vacuum-assisted systems) to prevent evisceration, manage fluid loss, and allow delayed closure once swelling resolves. Many consensus recommendations support decompression when pressures exceed 25 mm Hg, though some clinicians will intervene earlier in the right clinical context, recognizing that earlier decompression may improve outcomes.
Because mortality with ACS can be very high, often greater than 40%, patients require close inpatient monitoring—preferably in an intensive care unit—and early surgical consultation. The key clinical principle is that ACS is both a pressure problem and an organ failure syndrome: diagnosis requires evidence of intraabdominal hypertension plus end-organ dysfunction, and definitive management depends on timely decompression when supportive measures are insufficient.

- Published on
KembaraXtra-Medicine: Achilles Tendon Rupture
Achilles tendon rupture is a disruption of the continuity of the Achilles tendon, most commonly occurring after an eccentric loading event when the ankle is plantarflexed or when plantarflexion is forced against resistance. It is often an acute sports-related injury and can be missed initially, sometimes being mistaken for an ankle sprain. Relevant ICD-10-CM codes include S86.011 (right), S86.012 (left), M66.871 (nontraumatic right), M66.872 (nontraumatic left), and S86.0 (unspecified injury of Achilles tendon).
In the general population, Achilles tendon ruptures occur at an estimated rate of 18 per 100,000 people per year and are the most common large tendon rupture, accounting for approximately 20% to 35% of major tendon injuries. They most often occur in adults aged 30 to 55 years, with peak incidence in males aged 30 to 40 years, particularly those who participate in recreational athletics.
Risk is highest in people involved in “stop-and-go” sports such as basketball, tennis, and soccer, especially when there is a sudden increase in training intensity or duration. Additional risk factors include preexisting Achilles tendinopathy, advancing age, male sex, poor running mechanics, and a sedentary baseline followed by abrupt strenuous activity. Certain medications and substances are also associated with rupture risk, most notably fluoroquinolone antibiotics, where rupture tends to occur within the first 90 days, and steroid exposure, including prescribed corticosteroids and illicit anabolic steroid use.
The classic presentation is a middle-aged recreational athlete who feels a sudden “pop” in the posterior ankle or calf during pivoting, sprinting, jumping, or rapid deceleration, followed by acute pain and difficulty continuing activity. Pain may be severe, but some patients present primarily with weakness rather than pain and may still be able to walk. On examination, there may be swelling, bruising, and tenderness, and a palpable gap is often appreciated in the distal third of the tendon, typically 2 to 6 cm proximal to the calcaneal insertion. Patients may have an antalgic gait and are often unable to perform a single-leg heel raise due to loss of plantarflexion strength. When the patient lies prone with the feet hanging off the table, the injured side may rest in a more dorsiflexed position than the uninjured side. The Thompson test (calf squeeze test) is highly useful: squeezing the calf fails to produce normal plantarflexion when the tendon is ruptured. The Matles test (knee flexion test) provides additional support, with the injured foot falling into a more dorsiflexed or neutral position when the knees are flexed to 90 degrees.
Mechanistically, rupture is usually due to indirect trauma and reflects a combination of mechanical overload, relative hypovascularity in the classic “watershed zone” 2–6 cm above the insertion, and poor tendon tissue quality. Chronic tendinosis (degenerative change from repetitive microtears) and tendinitis can weaken the tendon, making it more susceptible to rupture during abrupt loading.
The differential diagnosis includes Achilles tendinopathy, retrocalcaneal bursitis, ankle sprain, calcaneal avulsion fracture, partial gastrocnemius tear, plantaris rupture, os trigonum syndrome, and calcaneal apophysitis. Diagnosis is largely clinical when history and exam findings are classic, but imaging can help in unclear cases or when partial tears are suspected.
Ultrasound is often used at the bedside and can identify tendon discontinuity, tendinopathy, and hematoma, making it a practical first-line study in many settings. MRI provides the greatest anatomic detail and is particularly helpful for confirming partial tears, defining the extent of injury, and assessing associated tendinopathy; a complete rupture typically shows fiber discontinuity and fluid signal at the tear site.
Initial management includes RICE (rest, ice, compression, elevation), along with analgesia such as acetaminophen or NSAIDs as appropriate. The ankle should be immobilized in a posterior slab splint with the foot in resting equinus (plantarflexion) to reduce tension on the tendon. Definitive treatment may be nonoperative or operative, and modern care increasingly uses accelerated functional rehabilitation protocols rather than prolonged casting alone.
Nonoperative treatment traditionally involved immobilization for 8 to 12 weeks, but newer accelerated protocols typically begin with about 2 weeks of immobilization, followed by progressive weight bearing in a walking boot with heel wedges to limit dorsiflexion, plus early supervised physical therapy emphasizing gradual range of motion and strengthening. These protocols have improved outcomes compared with older prolonged immobilization approaches.
Surgical repair is considered especially for acute complete ruptures, often aiming for repair within 4 weeks, and ideally within 7 to 14 days, to optimize healing and tendon length restoration. Techniques include open repair and percutaneous or minimally invasive approaches. Percutaneous repair may reduce operative time and deep infection risk but carries a higher risk of sural nerve injury. Postoperative care similarly relies on structured rehabilitation with early controlled motion and progressive strengthening.
Orthopedic referral is recommended for suspected acute or complete ruptures, as early evaluation helps guide operative versus nonoperative management and facilitates timely rehabilitation planning. Clinically important pearls include remembering that some patients may have minimal pain and may still walk, so careful palpation for a defect and performance of the Thompson test are crucial. Although surgery can reduce rerupture rates, it has higher complication risks, and randomized trials show that with modern functional rehabilitation, nonoperative treatment can achieve outcomes comparable to surgery at 12 months. Platelet-rich plasma (PRP), despite common use, has not shown benefit over placebo in several studies for acute rupture.
Prevention focuses on addressing modifiable risk factors, including maintaining conditioning, using proper training progression, continuing eccentric and concentric calf strengthening (especially after prior tendinopathy or rupture), and reducing training intensity during fluoroquinolone or anabolic steroid exposure.


- Published on
KembaraXtra-Medicine: Acetaminophen Poisoning
Acetaminophen (APAP) poisoning occurs when excessive amounts of acetaminophen are ingested, overwhelming normal hepatic metabolic pathways and leading to acute liver injury. If untreated, toxicity can progress to hepatic necrosis, liver failure, and death. Early symptoms are often nonspecific and include nausea, vomiting, diaphoresis, malaise, and abdominal discomfort, while later manifestations include jaundice, coagulopathy, hepatic encephalopathy, and multiorgan failure. Acetaminophen is also known as paracetamol, and poisoning is classified under ICD-10-CM codes T39.1 (accidental poisoning) and X60 (intentional self-poisoning).
Acetaminophen is among the most widely used antipyretic and analgesic medications worldwide and is present in over 100 prescription and over-the-counter combination products, including many cold, flu, and opioid formulations. In the United States alone, more than 100,000 potentially toxic ingestions are reported each year, making acetaminophen the most common pharmaceutical exposure reported to Poison Control Centers. It is the leading cause of acute liver failure and liver transplantation nationally. Nearly half of exposures occur in children younger than 6 years, often due to dosing errors. Individuals with chronic alcohol use disorder, malnutrition, chronic liver disease, or concurrent use of hepatotoxic drugs are at increased risk for severe toxicity.
The clinical course of acetaminophen poisoning typically follows four stages. During Stage I (0–24 hours), patients may be asymptomatic or experience nonspecific symptoms such as nausea, vomiting, anorexia, and lethargy, with laboratory studies often normal early on. Stage II (24–72 hours) marks the onset of hepatic injury, characterized by right upper quadrant abdominal pain, rising AST and ALT levels, and early abnormalities in coagulation or renal function. Stage III (72–96 hours) represents the period of maximal hepatotoxicity, with findings that may include hepatic encephalopathy, jaundice, coagulopathy, hypoglycemia, renal failure, metabolic acidosis, and death from multiorgan failure. Patients who survive this phase enter Stage IV (4 days to 2 weeks), during which hepatic regeneration occurs and clinical recovery is usually complete.
Acetaminophen toxicity results from the accumulation of N-acetyl-p-benzoquinone imine (NAPQI), a toxic metabolite normally detoxified by glutathione. When excessive acetaminophen is ingested, glutathione stores are depleted, allowing NAPQI to cause hepatocellular damage. The recommended maximum daily dose is 3-4 g in adults and 90 mg/kg in children. A single ingestion of ≥7.5 g in adults or ≥150 mg/kg in children is considered potentially toxic. Massive ingestions, often defined as >30–50 g, can produce early metabolic acidosis, altered mental status, and rapid progression to liver failure.
Diagnosis is based on history, timing of ingestion, and laboratory evaluation. A serum acetaminophen concentration obtained 4 hours or more after ingestion is plotted on the Rumack-Matthew nomogram to estimate the risk of hepatotoxicity and guide treatment decisions. The nomogram is not reliable in cases of unknown ingestion time, delayed presentation (>24 hours), chronic or staggered overdoses, or extended-release formulations. Additional laboratory studies include AST, ALT, INR, bilirubin, serum glucose, renal function tests, and screening for co-ingestants. Pregnancy testing is required in women of childbearing age.
Management centers on rapid initiation of therapy. Consultation with a Poison Control Center is strongly recommended. Activated charcoal may be administered within 4 hours of ingestion if the patient presents early. The definitive treatment is N-acetylcysteine (NAC), which replenishes glutathione, enhances non-toxic metabolism of acetaminophen, and improves outcomes even when started after liver injury has begun. NAC should be administered whenever toxicity is suspected or cannot be reliably excluded.
NAC may be given intravenously over 21 hours or orally over 72 hours. Intravenous NAC provides predictable absorption and a shorter treatment course but carries a risk of anaphylactoid reactions, whereas oral NAC is effective but commonly causes nausea and vomiting. Therapy should be continued if acetaminophen levels remain detectable, liver enzymes are elevated, or coagulopathy persists. In rare, severe cases with extremely high acetaminophen levels, metabolic acidosis, or altered mental status, hemodialysis may be indicated, with continued NAC administration.
All patients with confirmed acetaminophen poisoning require hospital admission, often to an intensive care unit for close monitoring. When treated promptly, prognosis is excellent, with approximately 90% of patients recovering fully. Acute liver failure is uncommon in young children. Psychiatric evaluation is necessary following intentional ingestions, and patient education on appropriate dosing and awareness of acetaminophen-containing combination products is essential to prevent recurrence.

- Published on
KembaraXtra-Medicine: Acanthosis Nigricans
Acanthosis nigricans (AN) is a dermatologic condition characterized by dark, velvety, thickened skin that appears in a symmetric distribution, most commonly affecting intertriginous areas such as the axillae, neck, and groin. The condition is most often associated with insulin resistance, particularly in the setting of obesity and diabetes mellitus, but it can also occur less commonly due to underlying malignancy, medication effects, or congenital syndromes. AN itself is not dangerous, but it is an important cutaneous marker of systemic disease and warrants further evaluation.
AN is coded under ICD-10-CM L83. The reported prevalence varies widely across studies, but available data suggest that AN is more commonly observed among Black Americans, Native Americans, and Hispanic Americans compared with White or Asian Americans. This condition can affect individuals of all ages. Major risk factors include obesity, insulin resistance, type 2 diabetes, and malignancy, particularly gastrointestinal adenocarcinomas in cases of malignant or paraneoplastic AN.
Clinically, individuals with AN are often asymptomatic, though some may report pruritus or irritation. On examination, the affected skin appears hyperpigmented, thickened, and velvety, initially presenting as dry, rough, gray-brown to black patches. The most commonly involved sites include the axillae, groin, posterior and lateral neck, and popliteal fossae. In more extensive or severe cases, involvement of mucosal surfaces such as the lips, buccal mucosa, conjunctiva, and areolae may be seen. Rapid onset, extensive involvement, or mucosal disease should raise concern for an underlying malignancy.
The pathogenesis of AN is primarily related to hyperinsulinemia, which leads to direct and indirect stimulation of insulin-like growth factor (IGF) receptors on keratinocytes and fibroblasts. This stimulation promotes epidermal proliferation and dermal thickening. Other growth factor pathways, including epidermal growth factor receptor (EGFR) and fibroblast growth factor receptor (FGFR) signaling, may also play contributory roles. Mechanical factors such as friction and perspiration are thought to exacerbate AN, which helps explain its predilection for intertriginous regions. Clinically, AN is often categorized as benign (metabolic-related) or malignant/paraneoplastic, though additional subclassifications exist.
The diagnosis of AN is typically clinical, based on history and physical examination. From a dermatologic standpoint, conditions that may resemble AN include intertriginous granular parakeratosis, confluent and reticulated papillomatosis, Haber syndrome, Dowling–Degos disease, and reticulate acropigmentation of Kitamura. Because AN is frequently a manifestation of systemic disease, evaluation should also consider associated or underlying conditions such as prediabetes, diabetes, obesity, metabolic syndrome, polycystic ovary syndrome (PCOS), nonalcoholic fatty liver disease (NAFLD), chronic glucocorticoid exposure, hypertension, hyperlipidemia, and malignancy.
The workup of AN should be guided by clinical context and focuses on identifying underlying causes. Recommended laboratory tests include fasting blood glucose and hemoglobin A1c to assess for prediabetes or diabetes, fasting lipid panel for hyperlipidemia, and liver function tests to screen for fatty liver disease. In patients with features of hyperandrogenism or menstrual irregularities suggestive of PCOS, serum total testosterone and early morning 17-hydroxyprogesterone may be indicated. Imaging studies are not routinely required but may include transvaginal ultrasound when PCOS is suspected or CT/MRI if there is concern for malignancy based on clinical features such as sudden onset or rapid progression.
Management of AN centers on treating the underlying condition, which is the most effective approach. In cases related to obesity and insulin resistance, weight loss through diet and exercise is the cornerstone of therapy and often leads to gradual improvement or resolution of skin findings. If AN is medication-induced, discontinuation of the offending agent is recommended when feasible. Associated conditions such as diabetes, PCOS, hypertension, hyperlipidemia, or malignancy should be managed according to standard guidelines.
When AN persists despite optimal management of the underlying cause, adjunctive dermatologic treatments may be used for cosmetic or symptomatic relief. These include topical retinoids, vitamin D analogues, keratolytic agents, and, in more refractory cases, systemic retinoids or laser therapy. Referral to endocrinology is often helpful for management of metabolic disorders, and referral to a nutritionist can support sustainable lifestyle changes.
A key clinical pearl is that acanthosis nigricans is a sign, not a diagnosis. Its presence should prompt clinicians to search for and address the underlying disease process. Preventive strategies focus on maintaining healthy lifestyle habits, including balanced nutrition, regular physical activity, and routine primary care follow-up with appropriate metabolic and cancer screening, which can reduce the risk of developing AN and its associated conditions.


- Published on
KembaraXtra-Medicine: Achalasia
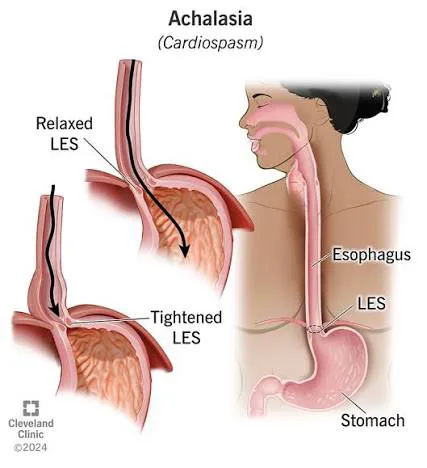
Achalasia is a chronic esophageal motility disorder characterized by impaired relaxation of the lower esophageal sphincter (LES) and absence of normal peristalsis in the smooth muscle portion of the esophagus. Together, these abnormalities create a functional obstruction at the esophagogastric junction, leading to progressive difficulty with esophageal emptying. Achalasia is treatable but not curable, and therapy focuses on relieving obstruction rather than restoring peristalsis. The condition is coded under ICD-10-CM K22.0 (achalasia of cardia) and is also referred to as cardiospasm, esophageal achalasia, aperistalsis of the esophagus, or megaesophagus.
The incidence of achalasia has increased with the widespread use of high-resolution esophageal manometry (HRM) and is currently estimated at 0.03–1.63 cases per 100,000 persons per year, with a prevalence of approximately 10 per 100,000. Although symptoms may begin at any age, achalasia demonstrates a bimodal age distribution, most commonly presenting between 20–40 years and again after 60 years of age. Men and women are affected equally, with a slightly higher incidence observed in older adults.
Patients most commonly present with progressive dysphagia to both solids and liquids, a key feature distinguishing achalasia from mechanical obstruction. Other frequent symptoms include regurgitation of undigested food, difficulty belching, chest pain or heartburn, globus sensation, frequent hiccups, nocturnal cough, aspiration symptoms, weight loss, and recurrent pneumonia. Disease severity and treatment response are often assessed using the Eckardt score, which evaluates dysphagia, regurgitation, chest pain, and weight loss. The Brief Esophageal Dysphagia Questionnaire (BEDQ) is a sensitive screening tool for dysphagia and may outperform symptom-based GERD scales. Physical examination is often benign, though lung findings such as wheezing or crackles may be present due to aspiration.
The precise etiology of achalasia remains incompletely understood. The disease is characterized by degeneration of inhibitory neurons in the esophageal myenteric plexus, leading to loss of nitric oxide and vasoactive intestinal polypeptide (VIP) signaling. This results in unopposed cholinergic excitation, incomplete LES relaxation, and aperistalsis. Histopathologic findings include inflammatory infiltrates, fibrosis, nerve hypertrophy, and capillaritis. Increasing evidence supports an autoimmune mechanism, with associations to specific HLA class II alleles, antineural antibodies, and a higher prevalence of other autoimmune diseases. Viral triggers, including herpes simplex virus and varicella zoster virus, have also been implicated. Secondary forms of achalasia may occur in Chagas disease, following vagal nerve injury, or after foregut surgery. Type III achalasia has been associated with chronic opioid use.
The differential diagnosis is broad and includes pseudoachalasia caused by esophageal or gastric malignancy, as well as other motility disorders such as diffuse esophageal spasm, scleroderma, gastroesophageal reflux disease, esophageal strictures, webs, rings, Barrett esophagus, and opioid-induced dysmotility. Non-esophageal causes such as angina, eating disorders, and gastric bezoars should also be considered.
Diagnostic evaluation typically includes endoscopy, barium esophagram, and esophageal manometry, which are complementary. Endoscopy is essential to exclude malignancy, strictures, and secondary causes of obstruction before manometry is performed. A timed barium esophagram may show classic findings such as esophageal dilation, retained food and secretions, poor emptying, and the characteristic “bird’s beak” tapering at the LES. In advanced disease, the esophagus may become tortuous and sigmoid-shaped.
High-resolution manometry is the gold standard for diagnosis and allows classification according to the Chicago Classification v4.0, which defines three achalasia subtypes. Type I (classic achalasia) shows complete aperistalsis with impaired LES relaxation. Type II includes aperistalsis with panesophageal pressurization and generally responds best to therapy. Type III (spastic achalasia) is characterized by premature or spastic distal esophageal contractions and often requires longer myotomy. HRM also identifies esophagogastric junction outflow obstruction (EGJOO), a related but distinct condition that requires correlation with symptoms and additional testing.
Treatment is aimed at lowering LES pressure to relieve obstruction and prevent progression to megaesophagus. Pneumatic dilation disrupts LES muscle fibers and provides symptom relief in many patients, though repeat dilations may be required and there is a small risk of perforation. Laparoscopic Heller myotomy, often combined with partial fundoplication, offers durable symptom control and is highly effective, though postoperative reflux is common. Peroral endoscopic myotomy (POEM) has emerged as an effective minimally invasive alternative, particularly for type III achalasia, but carries a higher risk of postprocedural reflux due to the absence of an antireflux barrier.
Medical therapy plays a limited role and is generally reserved for patients who are poor procedural candidates or require temporary symptom relief. Options include nitrates, calcium channel blockers, and botulinum toxin injections, the latter of which is particularly useful in elderly or medically frail patients but has limited durability. End-stage disease with severe dilation and failed prior therapy may require esophagectomy, though this is uncommon.
Key clinical considerations include recognizing that medications offer only transient benefit, while pneumatic dilation, surgical myotomy, and POEM provide more durable symptom control. Patients with long-standing achalasia have an increased risk of esophageal squamous cell carcinoma, and treated patients may develop reflux-related complications, including Barrett esophagus. Routine surveillance endoscopy is not universally recommended, but long-term clinical follow-up is essential.
- Published on
KembaraXtra-Medicine: Acne Vulgaris
Acne vulgaris is a chronic disorder of the pilosebaceous unit caused by abnormal shedding of follicular epithelium that blocks the follicular canal and promotes inflammation. This process leads to the development of comedones, papules, pustules, nodules/cysts, and, in some patients, scarring and dyspigmentation. Lesions are broadly categorized as noninflammatory (open and closed comedones) and inflammatory (papules, pustules, nodules, and cysts). Inflammatory acne may be described as papulopustular, nodulocystic, or mixed. Although many grading scales exist, there is no universally accepted severity system; clinically, acne is often described as mild, moderate, or severe based on lesion type and burden.
In general, mild acne is dominated by comedones with few inflammatory lesions (often fewer than 10) and no nodules or cysts. Moderate acne involves more numerous inflammatory papules and pustules (often 10–40) along with comedones, and “moderately severe” acne includes higher inflammatory counts or a small number of deeper nodulocystic lesions. Severe acne includes extensive papules and pustules and/or many nodulocystic lesions, often with early scarring. A pattern of deeper lesions along the mandible and anterolateral neck, menstrual flares, and signs of androgen excess such as hirsutism or irregular menses suggests hormonally driven acne.
Acne is commonly referred to simply as acne and is coded under ICD-10-CM L70.0 (acne vulgaris), with additional codes for specific variants such as acne conglobata, infantile/neonatal acne, acne excoriée, and others. Acne is the most common skin disease in the United States and is most prevalent in teenagers, affecting up to 85% to some degree. Incidence peaks between ages 15 and 18 in all genders. While acne often improves before age 25, a substantial minority persists into adulthood, especially in women, with a portion continuing into the mid-40s. Increased “acne mechanica” related to prolonged mask use has also been observed.
Clinical findings include open comedones (blackheads) and closed comedones (whiteheads), inflammatory papules and pustules, deeper nodules or cysts, and occasionally ectatic pores. Many patients develop postinflammatory erythema or hyperpigmentation, and some develop scars such as ice-pick, rolling, boxcar/crateriform, or keloidal scars. Lesions often coexist at different stages of evolution. The typical distribution is the face, upper chest, and back, while hormonal patterns more often involve the jawline and neck.
The underlying pathogenesis centers on comedone formation driven by increased sebum production and abnormal keratinization, which blocks follicles. This environment allows overgrowth of Cutibacterium acnes (formerly Propionibacterium acnes), promoting inflammation. Acne can be worsened by environmental factors such as hot, humid climates; certain medications and exposures (including iodine-containing cough mixtures, pomades/hair greases, anabolic steroids, progestins, lithium, and halogenated hydrocarbons); mechanical friction from sports gear or excessive washing; and stress, which may exacerbate acne via hypothalamic–pituitary–adrenal axis effects and androgen activation.
Diagnosis is clinical, but the differential is broad and includes folliculitis (including gram-negative), perioral dermatitis, rosacea, steroid acne, hidradenitis suppurativa, pseudofolliculitis barbae, keratosis pilaris, seborrheic dermatitis, drug eruptions, and occupational oil/grease exposure. Workup relies on history and physical examination, including prior treatments, skincare products, cosmetic use, family history, medication review (including OTC agents), and in females, the presence of cyclic flares or hormonal features. Clinicians should consider hyperandrogenism in women with hirsutism, irregular menses, or androgenic alopecia, and in children with early-onset acne, seborrhea, or acanthosis nigricans.
Routine laboratory testing is not typically needed. Patients who may be treated with isotretinoin should have baseline liver enzymes and triglycerides checked, and pregnancy testing is required before starting and during therapy due to strong teratogenic risk. If hyperandrogenism is suspected, evaluation may include DHEAS, total/free testosterone, LH, and FSH, though women with regular menses often do not require serum androgen testing.
Treatment is best approached with combination therapy targeting multiple pathogenic pathways. Nonpharmacologic options include selected light-based therapies such as blue light for moderate inflammatory acne, though long-term benefit compared with standard therapy is uncertain. Dietary counseling is appropriate in obese patients, and some evidence suggests high glycemic diets and certain dairy patterns may worsen acne, though the strength of association is debated.
For mild acne, first-line therapy typically includes benzoyl peroxide, a topical retinoid, or combination topical regimens, including topical antibiotics when inflammation is present. Topical therapies require 6 to 8 weeks of consistent use to assess effectiveness. Comedonal acne responds best to topical retinoids (e.g., adapalene, tretinoin, tazarotene), which normalize follicular keratinization and are comedolytic; salicylic acid washes can be helpful as adjuncts. Benzoyl peroxide is often added to reduce bacterial burden and inflammation but may cause irritation and dryness.
For inflammatory acne, topical antibiotics (such as clindamycin or erythromycin) are commonly used with benzoyl peroxide to reduce resistance and improve efficacy, and fixed-dose combination products can simplify regimens. Moderate to severe inflammatory acne may require oral antibiotics (commonly doxycycline or minocycline), ideally limited to 3–6 months to reduce resistance and microbiome disruption. Patients with nodulocystic acne or moderate-to-severe acne unresponsive to standard therapy may need systemic options such as isotretinoin, hormonal therapy (combined oral contraceptives), and/or antiandrogen therapy such as spironolactone; intralesional corticosteroid injections can rapidly improve large nodules.
Referral to dermatology is appropriate for severe, scarring, refractory acne or when procedures are needed, including intralesional injections, comedone extraction, and treatments for scarring such as resurfacing lasers, dermabrasion, chemical peels, or other scar-directed interventions. Important clinical pearls include recognizing that acne may worsen during the first few weeks of retinoid therapy before improving, and that worsening inflammatory acne after prolonged antibiotic use should raise suspicion for gram-negative folliculitis. Patients should be counseled that acne is usually controllable but not quickly “cured,” and meaningful improvement generally requires at least 4–8 weeks of consistent therapy, with attention to adherence and gentle skin care to minimize irritation and scarring.

