- Published on
KembaraXtra-Emergency and Acute Medicine - Optic Neuritis
Description: Optic neuritis is an inflammatory disorder of the optic nerve resulting in demyelination and acute optic nerve dysfunction. It is strongly associated with multiple sclerosis and is the presenting manifestation in approximately 15–20% of patients with MS. Inflammation may involve the optic disc (papillitis) or the retrobulbar portion of the optic nerve, where the funduscopic examination may initially appear normal. The long-term risk of developing clinically definite MS depends on MRI findings, with significantly higher risk in patients demonstrating multiple demyelinating lesions.
Etiology and risk factors: Most cases are idiopathic and self-limited, but 20–50% are associated with multiple sclerosis. Other causes include postviral inflammation following infections such as varicella, measles, mononucleosis, HSV, or VZV, typically occurring weeks after illness. Granulomatous and infectious causes include tuberculosis, syphilis, sarcoidosis, cryptococcosis, Lyme disease, and HIV-related infections. Drug-induced optic neuritis has been reported with amiodarone, ethambutol, and tamoxifen. Genetic predisposition is suggested by associations with HLA-A23, B7, and DR2 alleles.
Clinical features: Patients typically present with subacute vision loss developing over days, peaking within 1–2 weeks, most often unilateral in adults and bilateral in children. Retrobulbar pain, worsened by eye movement, is characteristic. Color vision, contrast sensitivity, and depth perception are disproportionately affected compared with visual acuity. An afferent pupillary defect is common in unilateral cases. Visual field testing often reveals a central scotoma. Funduscopic examination may show optic disc swelling or appear normal. Uhthoff phenomenon, transient worsening of vision with heat or exertion, may occur.
Evaluation: A detailed history should assess age, sex, onset and progression of visual loss, eye pain, prior neurologic symptoms, recent infections, drug exposure, and family history of MS. Physical examination requires a complete ophthalmologic and neurologic assessment including visual acuity, pupillary reflexes, color vision testing, visual fields, and dilated fundus examination. Blood pressure should be assessed to exclude hypertensive optic neuropathy.
Diagnostic testing: MRI of the brain and orbits with gadolinium is the imaging modality of choice and demonstrates optic nerve enhancement in the majority of acute cases while also stratifying future MS risk. CT is less sensitive and primarily used to exclude compressive lesions. Laboratory evaluation is guided by clinical suspicion and may include CBC, ESR, syphilis serology, Lyme testing, ANA, HIV testing, and tuberculosis screening. Chest radiography may assist in evaluating sarcoidosis or tuberculosis. Formal automated visual field testing is recommended for baseline assessment and follow-up.
Management: Early ophthalmology and neurology consultation is essential. High-dose IV corticosteroids followed by an oral taper are recommended for patients with severe visual loss or those with two or more demyelinating lesions on MRI, as this shortens recovery time and reduces short-term risk of MS progression. Oral corticosteroids alone should be avoided, as they increase recurrence risk. Treatment decisions should be individualized in patients with fewer MRI lesions.
Disposition and follow-up: Admission is indicated for bilateral vision loss, diagnostic uncertainty, or when IV steroid therapy is required. Patients with unilateral involvement, stable condition, and reliable follow-up may be discharged with urgent neurology and ophthalmology review. High-risk patients should be referred for disease-modifying therapy consideration. Prompt follow-up is mandatory, as MRI findings are the strongest predictor of future multiple sclerosis.
Key points: Space-occupying lesions must be excluded before diagnosing optic neuritis. Acute bilateral visual loss with headache or diplopia raises concern for alternative emergencies such as pituitary apoplexy. MRI is critical for prognostication, and management should be coordinated with specialists to align with current standards of care.

- Published on
KembaraXtra-Emergency and Acute Medicine - Organophosphate Poisoning
Description: Organophosphate poisoning results from irreversible inhibition of cholinesterase enzymes, particularly acetylcholinesterase, leading to accumulation of acetylcholine at central and peripheral synapses and subsequent cholinergic overdrive. Toxic effects involve muscarinic, nicotinic, and central nervous system pathways, which may overlap in presentation. Mortality is primarily due to respiratory failure caused by bronchorrhea, bronchoconstriction, respiratory muscle weakness, and central respiratory depression. In children, symptoms may be difficult to distinguish, and seizures occur more frequently than in adults.
Etiology: Exposure commonly occurs through agricultural insecticides or chemical nerve agents such as sarin, soman, tabun, and VX. These agents are rapidly absorbed through the lungs, gastrointestinal tract, skin, mucous membranes, and eyes, making even dermal exposure potentially life-threatening.
Clinical features: The classic presentation is the cholinergic toxidrome characterized by DUMBELS: diarrhea and diaphoresis, urination, miosis and muscle fasciculations, bradycardia with bronchorrhea and bronchospasm, emesis, lacrimation, and salivation. Mild exposure causes headache, dizziness, weakness, tremors, and anorexia. Moderate exposure leads to muscle fasciculations progressing to flaccid paralysis, respiratory muscle weakness, agitation, confusion, pinpoint pupils, nausea, vomiting, and excessive secretions. Severe exposure presents with seizures, coma, centrally mediated respiratory depression, bronchoconstriction, cyanosis, cardiac conduction abnormalities, profuse secretions, and urinary or fecal incontinence.
Evaluation: A focused history should assess occupational exposure, recent insecticide use, possible suicide attempt, and access to pesticides, with retrieval of the original container if available. Physical examination emphasizes identification of parasympathetic signs, muscle weakness, and respiratory compromise. Diagnosis is clinical, and treatment should not be delayed for laboratory confirmation.
Diagnostic testing: Red blood cell cholinesterase levels best reflect synaptic inhibition but are often delayed, while plasma cholinesterase levels are more rapidly available but less specific. Severity correlates with the degree of enzyme inhibition, though therapy must begin immediately regardless of results. Additional testing includes CBC, electrolytes, renal function, glucose, arterial blood gas when respiratory symptoms are present, ECG for dysrhythmias or heart block, and chest radiography if pulmonary edema or aspiration is suspected.
Management: Initial management prioritizes decontamination and protection of healthcare workers using appropriate personal protective equipment. All contaminated clothing should be removed and double-bagged, and skin thoroughly washed with soap and water. Airway protection, oxygenation, and ventilatory support are critical, with early intubation when indicated. Atropine is the primary antidote for muscarinic symptoms and should be administered in escalating doses every five minutes until bronchial secretions are dry, without using pupil size or heart rate as treatment endpoints. Pralidoxime should be given early to reverse nicotinic effects and regenerate cholinesterase before enzyme aging occurs, improving muscle strength and reducing paralysis. Supportive care includes frequent suctioning, avoidance of succinylcholine during intubation, and cautious gastric decontamination in early severe ingestions.
Disposition and follow-up: Any symptomatic patient or those requiring atropine should be admitted, typically to an intensive care setting, for close monitoring. Asymptomatic patients may be discharged after 6–12 hours of observation with clear return precautions and reliable follow-up. Intentional exposures require psychiatric evaluation, and poison control or toxicology consultation is recommended for significant or ongoing symptoms.
Key points: Inadequate atropine dosing is the most common cause of treatment failure. Clinical diagnosis should guide therapy without delay for laboratory confirmation. Recognition and aggressive management of respiratory compromise are essential to reduce mortality.

- Published on
KembaraXtra-Emergency and Acute Medicine - Osgood–Schlatter Disease
Description: Osgood–Schlatter disease is the most common cause of knee pain in children aged 10–15 years and represents a benign, self-limited extra-articular condition. It is characterized by pain, swelling, and tenderness over the tibial tuberosity at the insertion of the patellar tendon, just below the knee joint. Symptoms are activity related, worsening with exercise and improving with rest, and are commonly seen in physically active adolescents during periods of rapid growth.
Etiology: The most widely accepted mechanism involves repetitive traction and microfractures at the tibial tubercle apophysis caused by repeated stress from the patellar tendon. Activities that involve running, jumping, and sudden changes in direction increase strain on the extensor mechanism and precipitate symptoms.
Clinical features: Patients present with localized pain and swelling over the tibial tuberosity that is exacerbated by running, jumping, kneeling, or climbing stairs and relieved by rest. The condition is usually unilateral, although bilateral involvement occurs in approximately 20% of cases. Risk factors include age between 10 and 15 years, male sex, pubertal growth spurts, and participation in sports such as soccer, basketball, volleyball, and skating.
Physical examination: Examination reveals prominence, tenderness, and soft tissue swelling over the tibial tuberosity with pain reproduced by resisted knee extension. Quadriceps and hamstring tightness is common compared with the unaffected side. Mild erythema may be present, but the knee joint examination itself is otherwise normal, with no effusion or instability.
Evaluation: Diagnosis is primarily clinical based on history and examination. Imaging is not routinely required but may be obtained if the diagnosis is uncertain or to exclude other pathology. Plain knee radiographs may show fragmentation or irregular ossification of the tibial tuberosity, while ultrasound can demonstrate associated soft tissue changes.
Management: Treatment is conservative and focuses on symptom control and activity modification. Patients should rest from painful activities for approximately 6–8 weeks, particularly avoiding jumping and cutting sports. Ice application, stretching of the quadriceps and hamstrings, and use of analgesics such as ibuprofen or acetaminophen are recommended. An infrapatellar tendon strap or protective padding may reduce strain during activities. Corticosteroid injections should be avoided, and reassurance is essential as the condition resolves with skeletal maturity.
Disposition and follow-up: Admission is not required, and patients can be safely discharged home. Follow-up with a pediatrician or primary care provider in 2–3 weeks is advised to reassess symptoms and activity tolerance. Referral to pediatric orthopedics is rarely necessary and is reserved for patients

- Published on
KembaraXtra-Emergency and Acute Medicine - Osteogenesis Imperfecta
Description: Osteogenesis imperfecta is an inherited disorder caused by abnormalities in the procollagen amino acid sequence, leading to defective collagen formation. This results in bone hypomineralization, incomplete ossification, and brittle bones. Because collagen is a major component of connective tissue, multiple organ systems may be affected. The clinical course is variable, with most patients experiencing recurrent fractures during childhood followed by relative quiescence during adolescence and early adulthood.
Etiology: The condition arises from defects in procollagen that disrupt the structure of bone and connective tissue matrix. Mutations at different sites on the procollagen protein chain produce varying disease severity. Inheritance patterns include autosomal recessive forms, which are generally milder, and autosomal dominant forms, which are typically more severe. Lethal variants often result from sporadic or new mutations. Related disorders such as Ehlers–Danlos syndrome involve mutations in the same collagen protein but at different locations.
Clinical features: The hallmark manifestation is recurrent fractures, often occurring with minimal or trivial trauma, particularly in long bones. Fractures may be present at birth, recur throughout childhood, or reappear in the elderly. Skeletal abnormalities include bowed or shortened limbs, pectus excavatum, scoliosis, kyphosis, vertebral compression fractures, and abnormal skull shape. Blue sclerae are a classic finding and are not associated with visual impairment. Hearing loss, usually sensorineural, commonly begins in adolescence, with most patients affected by early adulthood. Dental abnormalities such as discolored, fragile, or misshapen teeth are frequent. Joint laxity, valvular disease, vascular abnormalities, and occasional thyroid dysfunction may also occur. Severe cases may result in perinatal death.
Pediatric considerations: In children, repeated fractures or fractures from low-impact mechanisms should raise suspicion for osteogenesis imperfecta. However, nonaccidental trauma must always be considered, and a careful social history and thorough evaluation are essential to distinguish between the two.
Evaluation: Diagnosis is typically based on a combination of clinical findings and radiographic features. A history of multiple fractures or fractures inconsistent with the reported mechanism is suggestive. A careful physical examination should assess for tenderness at other sites, blue sclerae, dental abnormalities, joint laxity, and neurovascular status distal to fractures.
Investigations: Laboratory studies are used to exclude metabolic causes of bone fragility, including abnormalities in calcium, phosphate, vitamin D, vitamin C, or parathyroid hormone levels. Genetic testing may be performed for confirmation, family counseling, or prenatal diagnosis. Radiographs often demonstrate osteopenia, crumpled or bowed long bones, incomplete ossification at physes, and characteristic findings such as wormian bones of the skull. A skeletal survey is mandatory in children. Audiologic testing should be arranged in older patients.
Management: There is no definitive cure for osteogenesis imperfecta. Acute management in the emergency setting focuses on stabilization, pain control, and appropriate immobilization of fractures. Fracture management is determined by injury type and location, with early orthopedic consultation for consideration of traction or operative fixation when indicated.
Disposition and follow-up: Admission is based on injury severity, presence of multiple fractures, or need for operative intervention. Pediatric patients may require admission to evaluate for possible nonaccidental trauma. Patients with isolated fractures and adequate home support may be managed as outpatients with close orthopedic and primary care follow-up. Long-term management focuses on monitoring disease progression, preventing fractures, and addressing hearing and mobility issues.
Key points: Osteogenesis imperfecta should be suspected in patients, especially children, with recurrent fractures or fractures from minor trauma. Differentiating pathologic fractures from nonaccidental trauma is critical and often challenging. Pain perception is normal in these patients, and respiratory infections may occur more frequently due to chest wall abnormalities.


- Published on
KembaraXtra-Emergency and Acute Medicine - Osteomyelitis
Description: Osteomyelitis is an infection of bone characterized by ongoing inflammatory destruction. It is most commonly bacterial in origin, although fungal osteomyelitis can occur, particularly in immunocompromised patients. The disease may present as acute, subacute, or chronic infection, with chronic cases defined by persistence or recurrence and the presence of necrotic bone (sequestrum).
Etiology: Hematogenous osteomyelitis occurs when bacteria seed bone via the bloodstream and is most common in children, the elderly, and people who inject drugs. Children typically develop acute disease, often without a preceding illness, though up to one-third report recent trauma. Adults more often develop subacute or chronic disease. Staphylococcus aureus is the most common pathogen across all age groups. Neonates are additionally affected by Enterobacteriaceae, group A and B streptococci, and Escherichia coli. Children may also develop infection from Haemophilus influenzae, while Salmonella is classically associated with sickle cell disease. Adults may have infections caused by gram-negative rods, Pseudomonas, Staphylococcus epidermidis, and anaerobes. Vertebral osteomyelitis is uncommon but typically affects adults over 45 years, often in the setting of diabetes, malignancy, hemodialysis, long-term catheterization, or IV drug use, and may extend to cause epidural abscesses or deep paraspinal collections. Direct or contiguous osteomyelitis occurs following trauma, open fractures, surgery, or spread from adjacent soft tissue infection and is more common in adults and adolescents. Chronic osteomyelitis is associated with necrotic bone and commonly involves S. aureus, S. epidermidis, Pseudomonas aeruginosa, and gram-negative organisms.
Clinical features: Symptoms vary with disease duration. Patients often present with localized, deep, dull, or throbbing bone pain that may occur at rest or with movement. Fever and chills may be present in acute disease but are often absent in chronic infection. Other features include malaise, nausea, vomiting, reluctance to use an affected limb, nonhealing ulcers, or fracture nonunion. Risk factors include diabetes mellitus, vascular disease, IV drug use, trauma, and invasive procedures. Examination may reveal localized warmth, erythema, edema, tenderness, decreased range of motion, sinus tract drainage, or exposed bone. Deep ulcers with palpable bone and a positive “probe-to-bone” test strongly suggest osteomyelitis.
Evaluation: Initial workup includes complete blood count, erythrocyte sedimentation rate, C-reactive protein, plain radiographs, and blood and wound cultures. Leukocytosis may be absent, but inflammatory markers are usually elevated. Blood cultures are positive in approximately half of cases. Plain radiographs are often normal in the first two to three weeks; early findings include periosteal elevation, followed by cortical erosion and new bone formation. MRI is the imaging modality of choice, with high sensitivity and specificity, allowing early detection and assessment of marrow, cortical, and soft tissue involvement. CT is useful when MRI is contraindicated and for surgical planning. Bone scans and leukocyte scintigraphy may be helpful in selected cases, while ultrasound is increasingly useful in children. Definitive diagnosis is established by bone biopsy with histology and culture, which remains the gold standard.
Management: Initial management focuses on stabilization, particularly in septic patients or those with neurologic deficits from spinal involvement. Empiric intravenous antibiotics should be started after cultures are obtained, then tailored based on organism and sensitivities. Treatment typically requires four to six weeks of parenteral antibiotics, with shorter IV courses followed by oral therapy possible in selected pediatric cases. Orthopedic and infectious disease consultation is essential, and surgical intervention is often required for debridement of necrotic bone, infected hardware, or abscesses.
Disposition and follow-up: Patients with acute osteomyelitis should be admitted for intravenous antibiotics and monitoring. Chronic osteomyelitis often requires admission for surgical management and prolonged therapy. Selected subacute or chronic cases may be managed as outpatients if debridement has been performed, cultures obtained, and reliable home IV antibiotic therapy is available. Close follow-up is mandatory to monitor response and prevent recurrence.
Key points: A normal white blood cell count does not exclude osteomyelitis. Early radiographs may be normal, making MRI critical for early diagnosis. Wound cultures alone are often unreliable for guiding therapy, and bone biopsy provides the most accurate microbiologic diagnosis.


- Published on
Emergency and Acute Medicine – Osteoporosis
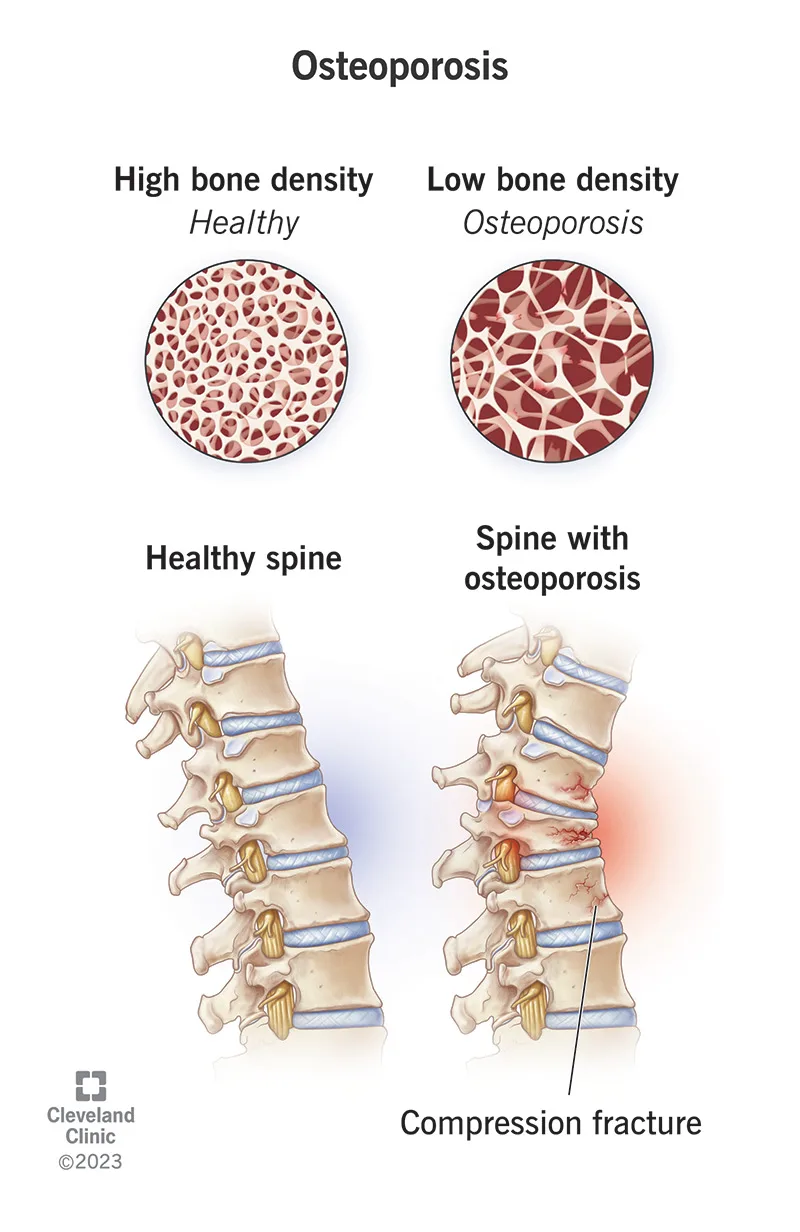
Osteoporosis is characterized by a diffuse decrease in skeletal mass, with trabecular bone—particularly in the vertebrae and femur—affected earlier and more severely than cortical bone. Although the disease process begins in adolescence, fractures typically do not manifest until age 50 years or older. Females are affected far more commonly than males, especially following menopause.
The underlying mechanism is an imbalance between bone resorption and bone formation, with resorption predominating. Advanced age is the most significant risk factor. Additional contributors include inadequate dietary calcium intake, particularly early in life, a sedentary lifestyle with lack of weight-bearing activity, and estrogen deficiency after menopause. Other important risk factors include long-term corticosteroid use, alcoholism, methotrexate therapy, tobacco use, low body weight, and familial predisposition. Pediatric patients are generally asymptomatic despite early onset of bone loss.
Osteoporosis is usually clinically silent until pathologic fractures occur. Fractures resulting from minimal trauma or recurrent fractures are hallmark features. The vertebral column is most commonly involved, with multiple compression fractures often leading to kyphosis and scoliosis. Hip fractures, including femoral neck and intertrochanteric fractures, are also frequent. A history of fractures after low-energy mechanisms or a family history of osteoporosis should raise suspicion. Physical examination findings typically relate to the acute fracture rather than the underlying disease itself.
Evaluation focuses on identifying fractures without significant mechanisms and assessing risk factors. A careful neurovascular examination distal to femoral or other extremity fractures is essential. In patients with vertebral fractures, rectal tone and postvoid residual should be assessed. Plain radiographs of suspected fractures may show osteopenia, though this is a late finding. Spine films may reveal old compression fractures. Computed tomography is recommended for vertebral fractures to assess for retropulsion and spinal canal compromise, which may not be evident on plain films.
Laboratory studies such as serum calcium, parathyroid hormone, and alkaline phosphatase can help differentiate osteoporosis from other metabolic bone disorders. Imaging with plain radiographs identifies fractures, but determining fracture age can be difficult. Bone scan or CT may help clarify fracture chronicity, particularly in the spine. Bone densitometry using dual-energy x-ray absorptiometry confirms the diagnosis, with a bone mineral density T-score of −2.5 or lower defining osteoporosis.
The differential diagnosis includes multiple myeloma or metastatic malignancy, osteogenesis imperfecta, hyperparathyroidism, and other demineralizing bone diseases. Management in the emergency setting prioritizes fracture care. Prehospital providers should minimize patient movement and report mechanism details suggestive of pathologic fracture. Initial treatment involves immobilization and standard fracture management, recognizing that healing may be delayed or incomplete. Orthotic back braces may be needed for vertebral fractures in consultation with orthopedics. Prevention remains more effective than acute treatment, and long-term therapy should be initiated or reinforced.
Pharmacologic therapy includes bisphosphonates such as alendronate or risedronate as first-line agents, with alternatives including zoledronic acid, raloxifene, denosumab, calcitonin, parathyroid hormone analogs, and, in selected cases, estrogen therapy. Adequate calcium and vitamin D supplementation are essential. Admission decisions follow standard orthopedic protocols, with particular attention to age, mobility, neurologic symptoms, and social factors. Compression fractures are often stable, but cervical fractures or those with neurologic deficits require admission and emergent specialty consultation.
Key points include recognizing recurrent or low-energy fractures as suggestive of osteoporosis, understanding that osteopenia on plain radiographs is a late but important clue, and initiating referral for definitive evaluation and treatment. Bisphosphonates remain first-line therapy for long-term management, and coordinated follow-up is critical to prevent future fractures.
- Published on
Emergency and Acute Medicine – Paget Disease
Paget disease, also known as osteitis deformans, is a disorder of bone remodeling characterized by excessive resorption of normal bone followed by replacement with disorganized, fibrous, and sclerotic bone. The disease is usually focal and most commonly affects the pelvis, femur, skull, tibia, and spine, particularly the lumbar spine. It occurs in approximately 1–2% of individuals older than 55 years, with incidence increasing with age. Many patients are asymptomatic, and the disease is often discovered incidentally on radiographs or through elevated alkaline phosphatase levels.
The disease begins with an osteolytic phase in which osteoclasts aggressively resorb healthy bone. This phase is associated with increased bone vascularity, predisposing patients to hematoma formation and pathologic fractures. Over time, the resorbed bone is replaced by dense, irregular trabecular bone in the osteoplastic phase, forming a characteristic mosaic pattern. Although malignant transformation is rare, occurring in about 1% of cases, osteosarcoma is the primary malignancy of concern. Paget disease is more common in men and individuals of European descent and is rare in children.
The etiology remains unclear, though both genetic and environmental factors are implicated. Mutations in the SQSTM1 gene have been identified in many cases, and viral inclusions seen in osteoclasts suggest a possible association with paramyxoviruses. Environmental factors such as rural living and exposure to farm animals may also contribute.
Clinical presentation varies widely. Many patients remain asymptomatic, while others develop deep, aching bone pain later in the disease course. Pain may worsen with weight bearing when the femur or tibia is involved or with rest when non–weight-bearing bones are affected. During the acute osteolytic phase, patients are at risk for pathologic fractures, hypercalcemia, renal stones, and bleeding due to hypervascular bone. In widespread disease, increased bone blood flow may lead to high-output cardiac failure. In the osteoplastic phase, long-bone involvement can cause deformity, gait abnormalities, and swelling. Skull involvement may lead to headaches, cranial nerve compression, hearing loss, or changes in head size, while spinal disease can cause neurologic compression.
Diagnosis is usually suggested by characteristic radiographic findings and supported by laboratory abnormalities. Alkaline phosphatase is the most sensitive marker of disease activity, while calcium and phosphate levels are typically normal unless complications such as fracture or immobilization occur. Imaging with plain radiographs often reveals lytic lesions, bone expansion, cortical thickening, or dense “ivory” bone. Radionuclide bone scans are useful to assess the extent and activity of disease, while CT or MRI helps evaluate complications such as neoplasm, hematoma, or spinal cord compression.
Emergency management focuses on complications rather than the underlying disease. Pathologic fractures require prompt immobilization to limit bleeding, and analgesia is provided with acetaminophen or opioids. Hypercalcemia is treated with intravenous fluids, calcitonin, and bisphosphonates as needed. Suspected neurologic compromise mandates urgent neurosurgical consultation. Definitive medical therapy is indicated for symptomatic patients or those with disease in high-risk locations and typically involves nitrogen-containing bisphosphonates such as alendronate, risedronate, pamidronate, or zoledronic acid.
Admission is indicated for patients with major trauma, significant bleeding, hypercalcemia, high-output cardiac failure, or neurologic compression. Stable patients with adequate pain control and no acute complications may be discharged with appropriate orthopedic and endocrinology follow-up. A key clinical pearl is that Paget disease is often an incidental diagnosis, and unexplained elevation of alkaline phosphatase or pathologic fracture in an older adult should prompt consideration of this condition.


- Published on
Emergency and Acute Medicine – Ovarian Cyst and Adnexal Torsion
Ovarian cysts are common gynecologic findings and are usually asymptomatic unless complicated by rupture, hemorrhage, infection, or torsion. The most frequent type is the follicular cyst, which can occur from fetal life through menopause. These cysts are typically thin-walled, unilocular, and measure between 3 and 8 cm. Because of their thin walls, they may rupture easily, often causing minimal bleeding and little pain. Midcycle rupture during ovulation is known as mittelschmerz and is generally benign.
Corpus luteal cysts are clinically more significant. They are usually smaller than 10 cm but are prone to intracystic hemorrhage. Rapid bleeding can lead to cyst rupture, most commonly just before the onset of menses, and may result in severe intraperitoneal hemorrhage. In some cases, gradual bleeding into the cyst or ovary causes capsular distension and pain even without rupture. Anticoagulated patients are at particularly high risk for significant bleeding from corpus luteal cysts.
Adnexal torsion is a true gynecologic emergency and represents the fifth most common surgical emergency in gynecology. It occurs when the ovary, fallopian tube, or a paratubal cyst twists around its vascular pedicle, leading to impaired lymphatic and venous drainage followed by arterial compromise. This process causes rapid adnexal enlargement, ischemia, and eventual necrosis if not promptly treated. The greatest risk of torsion is associated with ovarian cysts measuring 8–12 cm. Although torsion can occur at any age, it is most common in reproductive-age women, with approximately 15% of cases occurring in children. During pregnancy, torsion is most likely in the first trimester, especially following ovarian stimulation or in vitro fertilization.
Patients with ovarian cysts typically present with unilateral lower abdominal or pelvic pain that may be sharp or aching and intermittent or constant. Pain may be precipitated by exercise, intercourse, trauma, or pelvic examination. Fever is uncommon and suggests an alternative diagnosis. Menstrual irregularities, infertility, pregnancy status, and prior sexually transmitted infections should be assessed. In contrast, adnexal torsion often presents with sudden-onset, severe, colicky abdominal pain that may localize to one side or radiate to the groin or flank. Nausea and vomiting are common, and symptoms may wax and wane due to intermittent torsion and detorsion. Fever and vaginal bleeding may occur but are not universal.
Physical examination in ovarian cysts may reveal abdominal or adnexal tenderness and, occasionally, a palpable pelvic mass. In cases of hemorrhagic cyst rupture, signs of hypovolemia such as orthostasis, tachycardia, or hypotension may be present. Adnexal torsion similarly causes abdominal and adnexal tenderness and may be associated with a palpable mass. Severe cases can mimic other acute abdominal emergencies.
A pregnancy test is essential in all patients of reproductive age to exclude ectopic pregnancy. Initial laboratory evaluation includes a complete blood count to assess for anemia or infection and urinalysis to evaluate urinary causes of pain. If significant hemorrhage is suspected, type and crossmatching for packed red blood cells should be performed. Cervical cultures may be indicated if pelvic inflammatory disease is a concern.
Transvaginal ultrasound is the imaging modality of choice. It can identify ovarian cysts, adnexal masses, free pelvic fluid, and an enlarged, edematous ovary suggestive of torsion. Doppler flow studies may demonstrate decreased or absent blood flow, but the presence of flow does not exclude torsion, particularly if detorsion has occurred. MRI may be useful in pregnant patients with nondiagnostic ultrasound findings, while CT imaging can help identify alternative diagnoses or provide supportive evidence of torsion when ultrasound is inconclusive. Ultimately, laparoscopy remains the gold standard for diagnosing adnexal torsion and allows for definitive treatment.
The differential diagnosis includes ectopic pregnancy, pelvic inflammatory disease, appendicitis, endometriosis, round ligament pain, ovarian or metastatic neoplasm, and uterine torsion. In postmenopausal women, ovarian cysts are concerning for malignancy until proven otherwise.
Management depends on the diagnosis and clinical stability. Suspected adnexal torsion requires urgent gynecologic consultation and surgical intervention, as early detorsion improves the likelihood of ovarian salvage, particularly in pediatric patients. Ruptured ovarian cysts without significant hemorrhage may be managed conservatively with analgesia and close follow-up. Patients with ongoing pain, hemodynamic instability, or significant hemoperitoneum require admission and possible surgical management.
A key clinical pearl is that adnexal torsion is primarily a clinical diagnosis. Imaging studies may appear reassuring, but normal Doppler flow does not rule out torsion. Therefore, adnexal torsion should always remain high on the differential diagnosis in women and girls presenting with acute abdominal or pelvic pain.


- Published on
Emergency and Acute Medicine – Otologic Trauma
Otologic trauma involves injury to the external, middle, or inner ear and may result from blunt, penetrating, barotrauma, thermal, chemical, or blast mechanisms. Injuries to the pinna are particularly concerning because auricular cartilage lacks a direct blood supply and depends on the perichondrium for nutrition. Disruption of this relationship, especially from hematoma formation, can lead to ischemia, perichondritis, cartilage necrosis, and permanent deformity such as cauliflower ear. Penetrating injuries and bite wounds increase the risk of cartilage infection.
The middle ear is an air-filled cavity containing the ossicles and is vulnerable to pressure-related injuries such as blasts or diving accidents. Because it is bordered by the temporal and mastoid bones and adjacent to the cranial vault, fractures in this region may result in cerebrospinal fluid otorrhea or rhinorrhea and disruption of the vestibular system. The facial nerve traverses the middle ear, and injury can result in peripheral facial nerve paralysis. Pediatric patients with otologic trauma should always be evaluated for possible nonaccidental injury.
Patients may present with severe ear pain, bleeding, or visible deformity of the auricle. Auricular hematomas typically appear as bluish, fluctuant, or doughy swellings of the pinna and are associated with loss of normal contour. Lacerations, partial avulsions, or complete amputations may be seen. Signs of middle ear trauma include decreased hearing, tinnitus, vertigo, nystagmus, facial nerve weakness, and canal drainage. Partial hearing loss suggests tympanic membrane rupture, whereas complete hearing loss raises concern for ossicular chain or inner ear injury. Findings suggestive of basilar skull fracture include hemotympanum, Battle sign, and cerebrospinal fluid leakage from the ear or nose.
History should focus on the mechanism of injury, associated trauma, prior otologic disease, and medication use. Physical examination must include a complete head and neck evaluation with careful inspection of the pinna, external auditory canal, tympanic membrane, cranial nerves, and hearing assessment. Tuning fork tests such as Weber and Rinne are useful to differentiate conductive from sensorineural hearing loss. Concomitant injuries must always be excluded in trauma patients.
Diagnosis is primarily clinical. Wound cultures are reserved for signs of infection. Imaging with head or facial CT is indicated when intracranial injury or skull fracture is suspected, while CT of the temporal bone without contrast is recommended for significant middle ear injury or facial nerve involvement.
Initial management begins with standard trauma assessment and stabilization, including airway, breathing, and circulation. Injured ears should be covered with sterile dressings. If the auricle is amputated, it should be wrapped in moist gauze, placed in a plastic bag, and kept cool for possible reattachment. Adequate anesthesia is essential and may be achieved through auricular nerve blocks or a circumferential ring block at the base of the pinna.
Auricular hematomas require prompt drainage, ideally within 72 hours, to reapproximate the perichondrium and cartilage and prevent necrosis. Small, nonclotted hematomas may be aspirated, while larger or clotted collections are best managed with incision and drainage followed by a firm pressure dressing. Antistaphylococcal antibiotics are typically given for 7–10 days. Reaccumulation requires repeat drainage and possibly wick placement.
Lacerations should be thoroughly cleaned and debrided. Simple lacerations can be closed with nonabsorbable monofilament sutures and a pressure dressing, while exposed cartilage must be carefully covered to prevent perichondritis. Human or animal bite wounds warrant prophylactic antibiotics, most commonly amoxicillin–clavulanate. Small avulsions less than 2 cm may survive as grafts, but larger avulsions require urgent consultation with otolaryngology or plastic surgery.
Patients may be discharged if they can tolerate oral antibiotics, have no serious associated injuries, and can ensure close follow-up. Admission is indicated for patients with significant associated trauma, immunosuppression with infection, perichondritis, chondritis, or need for intravenous antibiotics. Follow-up is recommended within 24 hours for auricular hematomas to assess for reaccumulation and within five days for suture evaluation or removal.
A key clinical principle is that failure to promptly recognize and adequately treat auricular hematomas can result in permanent deformity. Early drainage, proper pressure dressing, and close follow-up are essential to prevent long-term complications.


- Published on
Emergency and Acute Medicine – Otitis Externa
Otitis externa is an inflammation or infection of the auricle, external auditory canal, or the external surface of the tympanic membrane, while sparing the middle ear. It affects approximately 4 per 1,000 individuals in the United States and is commonly referred to as “swimmer’s ear” due to its frequent association with recent swimming or water exposure, although it may also occur after routine bathing. A severe form, necrotizing (malignant) otitis externa, begins in the ear canal and can extend into periauricular tissues and the skull base. This form occurs most often in elderly patients, those with diabetes, or immunocompromised individuals, is commonly caused by Pseudomonas aeruginosa, and carries a mortality rate of up to 20%.
The condition is often precipitated by trauma or abrasion of the ear canal or by maceration of the skin from prolonged moisture or excessive dryness. Predisposing factors include prior ear surgery or tympanic membrane perforation, narrow or abnormal ear canals, humidity, allergies, eczema, trauma, and abnormal cerumen production. Common pathogens include Pseudomonas aeruginosa, Staphylococcus aureus, streptococcal species, and less commonly fungal organisms.
Patients typically present with a history of recent swimming or water exposure and may report itching of the ear canal as the earliest symptom. This is often followed over one to two days by progressive ear pain, ear drainage, decreased hearing, and a sensation of ear fullness or clogging. In patients with diabetes or immunosuppression, clinicians should specifically inquire about systemic symptoms and risk factors. On examination, pain is elicited with movement of the pinna or tragus, and the external ear canal is usually swollen, erythematous, and tender, often with visible drainage or debris. Decreased auditory acuity and preauricular swelling may be present. Necrotizing otitis externa may present with severe otalgia, headache, otorrhea, periauricular swelling, and cranial nerve palsies, most commonly involving the facial nerve.
Diagnosis is primarily clinical, based on characteristic history and physical findings. Otoscopic examination typically reveals an edematous, erythematous ear canal with purulent, cheesy white, or gray-green exudate. Laboratory studies are not routinely required unless necrotizing otitis externa is suspected, in which case evaluation may include white blood cell count, erythrocyte sedimentation rate, serum glucose, and cultures. Computed tomography or magnetic resonance imaging is indicated when there are signs of systemic toxicity, suspected bony involvement, or concern for mastoiditis. Debris removal from the ear canal using gentle curettage or irrigation is both diagnostic and therapeutic, and wick placement may be necessary to ensure medication penetration in markedly edematous canals.
The differential diagnosis includes otitis media, folliculitis, foreign bodies in the ear canal, herpes zoster oticus, parotitis, mastoiditis, dental abscess, sinusitis, temporomandibular joint disorders, and cervical adenitis. In children, an ear canal foreign body should be considered when purulent drainage is present.
Management focuses on pain control, eradication of infection, and prevention of recurrence. Treatment begins with gentle cleansing of the external auditory canal to remove debris, sometimes requiring suction or curettage. A cotton or gauze wick may be placed in cases of severe canal edema. Most cases respond well to topical therapy using antiseptic, anti-inflammatory, and antibacterial ear drops. Acetic acid solutions, combination antibiotic-steroid drops, or fluoroquinolone drops such as ofloxacin are commonly used, with ofloxacin preferred when tympanic membrane perforation is suspected. Oral antibiotics are reserved for patients with facial or neck cellulitis, severe canal edema, concurrent otitis media, or immunocompromised status. Diabetic and immunocompromised patients should be treated with oral ciprofloxacin and monitored closely for progression to necrotizing otitis externa. Intravenous antibiotics and possible surgical debridement are required for necrotizing disease or severe systemic illness.
Most patients can be safely discharged with close follow-up. Admission is indicated for necrotizing otitis externa, significant pinna involvement, or signs of systemic illness. Discharge instructions should emphasize keeping the ear dry, avoiding swimming for three to four weeks, proper use of prescribed medications, and returning promptly for worsening pain, fever, hearing loss, or neurologic symptoms. Follow-up within two to three days is recommended, particularly for wick removal or if symptoms fail to improve.
Key clinical pearls include recognizing that erythema of the tympanic membrane in otitis externa may mimic otitis media, avoiding ear canal lavage until tympanic membrane integrity is confirmed, and ensuring adequate medication penetration by clearing obstructing debris. Recurrence can often be prevented through patient education focused on minimizing moisture, trauma, and irritant exposure to the external ear canal. Necrotizing otitis externa should always be suspected in diabetics or immunocompromised patients with severe ear pain, purulent otorrhea, and granulation tissue or exposed bone in the ear canal.

