- Published on
KembaraXtra-Medicine – Asthma
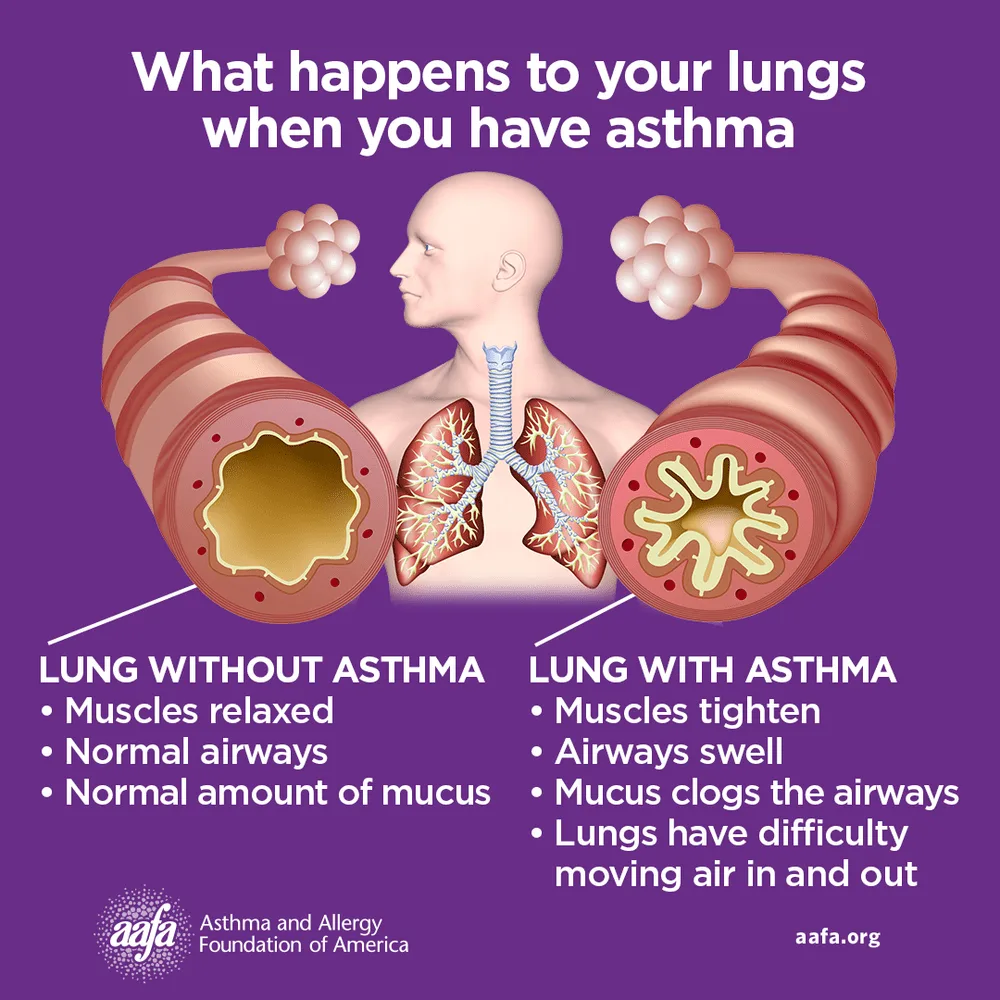
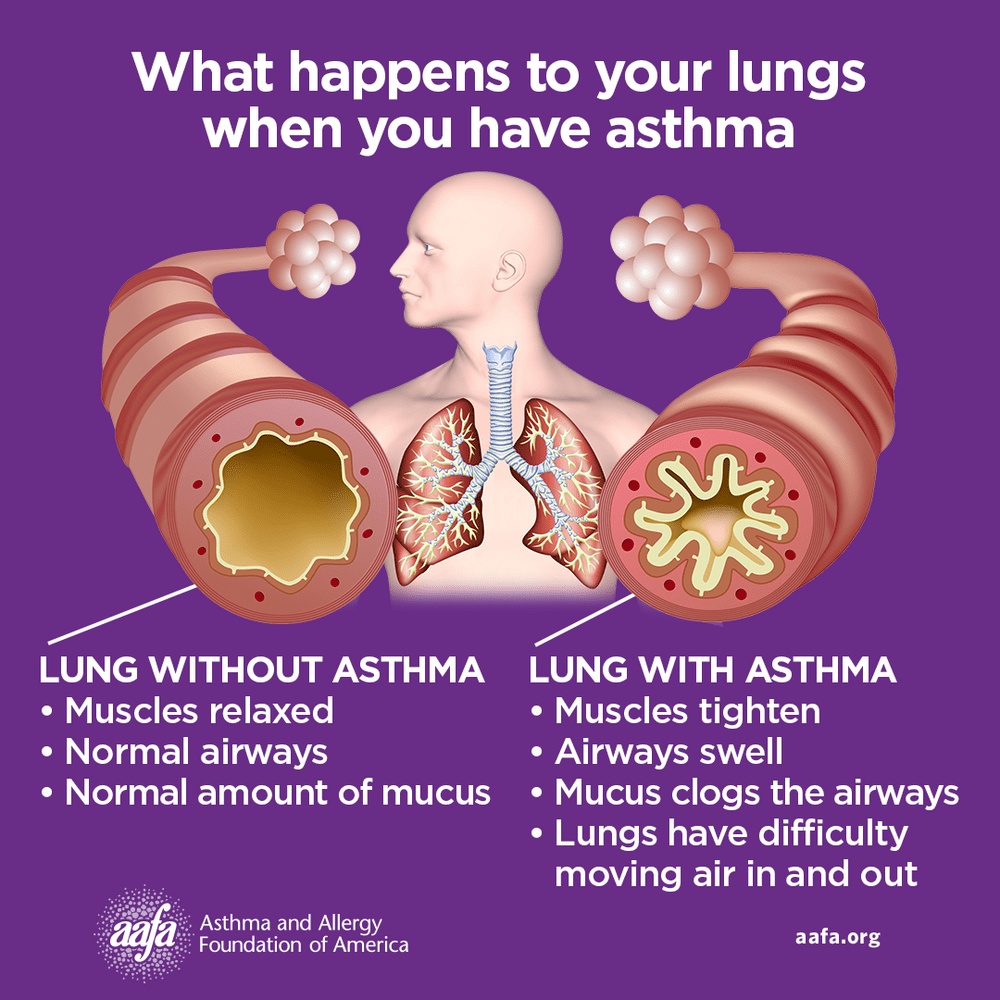
Asthma is a chronic inflammatory disease of the airways involving multiple cells, including mast cells, eosinophils, neutrophils, T lymphocytes, macrophages, and epithelial cells. This inflammation leads to recurrent episodes of cough (often at night or early morning), wheezing, shortness of breath, and chest tightness. These episodes are associated with variable and usually reversible airflow obstruction, either spontaneously or with treatment. Status asthmaticus refers to acute severe asthma that does not respond to standard therapies such as inhaled beta-agonists or subcutaneous epinephrine and may persist for hours, representing a life-threatening emergency.
Asthma is also referred to as bronchospasm, reactive airway disease, or asthmatic bronchitis. In the United States, approximately 7.7% of the population has asthma, with rising prevalence among adults, older individuals, women, African Americans, and people living below the poverty level. Globally, about 300 million people are affected, with projections reaching 400 million. Asthma accounts for significant healthcare use, including hundreds of thousands of hospitalizations and millions of emergency visits annually. Although mortality has declined overall, it remains unchanged in children aged 1–14 years. Many patients develop symptoms early in life, with 50–80% of children becoming symptomatic before age five.
Clinical presentation varies depending on severity and disease stage. Physical examination may be normal between attacks, but persistent or acute asthma often shows wheezing and prolonged expiration. Severe asthma and status asthmaticus may present with tachypnea, tachycardia, use of accessory muscles, pulsus paradoxus, altered mental status, paradoxical abdominal movement, or even a “silent chest,” which signals critical airflow obstruction. Risk factors for severe or fatal asthma include prior intubation, poor disease control, steroid dependence, smoking, obesity, psychiatric illness, advanced age, and limited access to medical care.
Asthma pathophysiology reflects a complex interaction between genetic susceptibility and environmental triggers. Allergic (atopic) asthma is driven by IgE-mediated responses to allergens, while nonallergic asthma often presents in adulthood following respiratory infections or stress. Occupational exposures, air pollutants, mold, exercise, medications such as NSAIDs or beta-blockers, and tobacco smoke can precipitate symptoms. Type-2 inflammatory pathways play a central role, involving cytokines such as IL-4, IL-5, and IL-13, leading to eosinophilia, IgE production, airway hyperresponsiveness, and remodeling. Genetic associations, including ADAM33 and other immune-related loci, further influence disease expression.
Diagnosis requires demonstration of airflow obstruction with reversibility. Spirometry before and after bronchodilator use is the preferred diagnostic test in patients older than five years, with reversibility defined as an increase in FEV₁ of at least 12% and 200 mL. Normal spirometry does not exclude asthma; bronchial challenge testing may be used when suspicion remains high. In young children, diagnosis is often clinical after exclusion of alternatives. Differential diagnoses include COPD, GERD, postnasal drip, vocal cord dysfunction, heart failure, pulmonary embolism, anxiety disorders, and interstitial lung disease.
Laboratory and ancillary testing help assess severity and guide management. Blood eosinophilia and elevated IgE support an allergic phenotype. Arterial blood gases are useful in acute severe attacks to assess hypoxia and hypercapnia. Imaging is usually normal but may show hyperinflation during exacerbations. Fractional exhaled nitric oxide and allergy testing can help phenotype asthma and predict response to therapy. Regular spirometry and peak flow monitoring are important for long-term assessment and self-management.
Management focuses on achieving and maintaining asthma control. Nonpharmacologic measures include trigger avoidance, smoking cessation, patient education, proper inhaler technique, and regular follow-up. Pharmacologic treatment follows a stepwise approach based on symptom frequency and severity. Short-acting beta-agonists are used for quick relief, while inhaled corticosteroids are the cornerstone of maintenance therapy. Additional controllers include long-acting beta-agonists (only in combination with inhaled corticosteroids), leukotriene receptor antagonists, long-acting muscarinic antagonists, and theophylline in selected cases. Therapy is stepped up when control is poor and stepped down when stable.
Patients with severe or refractory asthma may benefit from advanced therapies. Biologic agents targeting IgE, IL-5, IL-5 receptors, IL-4/IL-13 pathways, or thymic stromal lymphopoietin are used in carefully selected patients based on phenotype and biomarkers. Bronchial thermoplasty is an option for some adults with severe persistent asthma unresponsive to maximal medical therapy. Status asthmaticus requires aggressive management with oxygen, frequent bronchodilators, systemic corticosteroids, adjunctive agents such as magnesium sulfate, and ventilatory support when necessary.
Overall, asthma outcomes depend on individualized care, control of comorbid conditions, adherence to therapy, and early recognition of worsening symptoms. With appropriate management, most patients can achieve good symptom control, reduce exacerbations, and maintain normal activity levels.
Asthma is a chronic inflammatory disease of the airways involving multiple cells, including mast cells, eosinophils, neutrophils, T lymphocytes, macrophages, and epithelial cells. This inflammation leads to recurrent episodes of cough (often at night or early morning), wheezing, shortness of breath, and chest tightness. These episodes are associated with variable and usually reversible airflow obstruction, either spontaneously or with treatment. Status asthmaticus refers to acute severe asthma that does not respond to standard therapies such as inhaled beta-agonists or subcutaneous epinephrine and may persist for hours, representing a life-threatening emergency.
Asthma is also referred to as bronchospasm, reactive airway disease, or asthmatic bronchitis. In the United States, approximately 7.7% of the population has asthma, with rising prevalence among adults, older individuals, women, African Americans, and people living below the poverty level. Globally, about 300 million people are affected, with projections reaching 400 million. Asthma accounts for significant healthcare use, including hundreds of thousands of hospitalizations and millions of emergency visits annually. Although mortality has declined overall, it remains unchanged in children aged 1–14 years. Many patients develop symptoms early in life, with 50–80% of children becoming symptomatic before age five.
Clinical presentation varies depending on severity and disease stage. Physical examination may be normal between attacks, but persistent or acute asthma often shows wheezing and prolonged expiration. Severe asthma and status asthmaticus may present with tachypnea, tachycardia, use of accessory muscles, pulsus paradoxus, altered mental status, paradoxical abdominal movement, or even a “silent chest,” which signals critical airflow obstruction. Risk factors for severe or fatal asthma include prior intubation, poor disease control, steroid dependence, smoking, obesity, psychiatric illness, advanced age, and limited access to medical care.
Asthma pathophysiology reflects a complex interaction between genetic susceptibility and environmental triggers. Allergic (atopic) asthma is driven by IgE-mediated responses to allergens, while nonallergic asthma often presents in adulthood following respiratory infections or stress. Occupational exposures, air pollutants, mold, exercise, medications such as NSAIDs or beta-blockers, and tobacco smoke can precipitate symptoms. Type-2 inflammatory pathways play a central role, involving cytokines such as IL-4, IL-5, and IL-13, leading to eosinophilia, IgE production, airway hyperresponsiveness, and remodeling. Genetic associations, including ADAM33 and other immune-related loci, further influence disease expression.
Diagnosis requires demonstration of airflow obstruction with reversibility. Spirometry before and after bronchodilator use is the preferred diagnostic test in patients older than five years, with reversibility defined as an increase in FEV₁ of at least 12% and 200 mL. Normal spirometry does not exclude asthma; bronchial challenge testing may be used when suspicion remains high. In young children, diagnosis is often clinical after exclusion of alternatives. Differential diagnoses include COPD, GERD, postnasal drip, vocal cord dysfunction, heart failure, pulmonary embolism, anxiety disorders, and interstitial lung disease.
Laboratory and ancillary testing help assess severity and guide management. Blood eosinophilia and elevated IgE support an allergic phenotype. Arterial blood gases are useful in acute severe attacks to assess hypoxia and hypercapnia. Imaging is usually normal but may show hyperinflation during exacerbations. Fractional exhaled nitric oxide and allergy testing can help phenotype asthma and predict response to therapy. Regular spirometry and peak flow monitoring are important for long-term assessment and self-management.
Management focuses on achieving and maintaining asthma control. Nonpharmacologic measures include trigger avoidance, smoking cessation, patient education, proper inhaler technique, and regular follow-up. Pharmacologic treatment follows a stepwise approach based on symptom frequency and severity. Short-acting beta-agonists are used for quick relief, while inhaled corticosteroids are the cornerstone of maintenance therapy. Additional controllers include long-acting beta-agonists (only in combination with inhaled corticosteroids), leukotriene receptor antagonists, long-acting muscarinic antagonists, and theophylline in selected cases. Therapy is stepped up when control is poor and stepped down when stable.
Patients with severe or refractory asthma may benefit from advanced therapies. Biologic agents targeting IgE, IL-5, IL-5 receptors, IL-4/IL-13 pathways, or thymic stromal lymphopoietin are used in carefully selected patients based on phenotype and biomarkers. Bronchial thermoplasty is an option for some adults with severe persistent asthma unresponsive to maximal medical therapy. Status asthmaticus requires aggressive management with oxygen, frequent bronchodilators, systemic corticosteroids, adjunctive agents such as magnesium sulfate, and ventilatory support when necessary.
Overall, asthma outcomes depend on individualized care, control of comorbid conditions, adherence to therapy, and early recognition of worsening symptoms. With appropriate management, most patients can achieve good symptom control, reduce exacerbations, and maintain normal activity levels.
- Published on
KembaraXtra-Medicine – Asthma–COPD Overlap (ACO)
Asthma–COPD overlap (ACO) is a recognized clinical entity describing patients—often current or former smokers—who have chronic obstructive pulmonary disease (COPD) but also share important inflammatory and clinical features of asthma. Compared with COPD alone, ACO tends to be a more severe phenotype, with greater symptom burden and worse overall outcomes. ACO may be described as asthma with partially reversible airflow obstruction (with or without emphysema or reduced diffusing capacity) or as COPD with emphysema accompanied by reversible or partially reversible airflow obstruction (with or without allergies or reduced diffusing capacity). Because patients present with different patterns—such as COPD with eosinophilia, severe asthma in smokers, neutrophilic-predominant asthma, or asthma that has become largely irreversible due to airway remodeling—the term “ACO” is preferred over the older “ACOS” to reflect this heterogeneity.
ACO is discussed using overlapping ICD-10 codes including asthma (J45), chronic bronchitis (J42), emphysema (J43), COPD (J44, J44.9), and other overlap syndromes (M35.1). Epidemiologically, COPD prevalence varies across regions, and asthma affects tens of millions of people in the U.S. Bronchial hyperresponsiveness is common in COPD and bronchodilator reversibility can also be seen in COPD, which complicates diagnosis. Across studies, patients identified as having ACO generally demonstrate worse lung function, more respiratory symptoms, more frequent exacerbations, higher health-care utilization, and lower quality of life than those with asthma or COPD alone. Reported prevalence of ACO among adults with obstructive airway disease is commonly estimated around 15%–25%, with higher prevalence in older adults and in those with more severe disease. Some data suggest mortality in ACO is similar to COPD and worse than asthma alone.
Risk factors for ACO include cigarette smoking and atopy, along with older age, allergies, higher BMI, childhood asthma history, and respiratory infections such as rhinovirus or influenza. While no definitive genetic basis is established, some research has identified variants (including in GPR65) associated with ACO in certain populations. Clinically, patients may have wheezing, dyspnea, chest tightness, chronic cough (often productive), reduced exercise tolerance, recurrent respiratory infections, and episodic symptoms triggered by allergens, odors, or temperature changes. Physical examination can be normal or may show wheezing or rhonchi; in more severe cases, decreased air entry and accessory muscle use may appear, and signs such as the Hoover sign can suggest hyperinflation.
Diagnosis is considered when a patient demonstrates a mixed pattern of asthma-like and COPD-like features. A practical approach relies on objective confirmation of persistent airflow obstruction plus evidence of reversibility or airway hyperresponsiveness. Patients should have a reduced post-bronchodilator FEV₁/FVC (below the lower limit of normal or <0.7), along with either bronchodilator reversibility (increase in fev₁ or fvc by ≥200 ml and ≥12%) airway hyperresponsiveness demonstrated a positive methacholine challenge. because can occur copd symptoms overlap substantially, aco remains clinical diagnosis supported the pattern of symptoms, smoking history, asthma />llergy features, and pulmonary function testing rather than a single definitive test.
The differential diagnosis includes asthma, COPD, central airway obstruction, bronchiectasis, heart failure, and obliterative bronchiolitis. Workup typically includes spirometry with bronchodilator testing, and sometimes airway provocation testing. Additional supportive testing may include ABG (especially in severe disease), CBC with eosinophils, total IgE, sputum assessment, allergy testing, and peak flow monitoring. Imaging with chest x-ray or chest CT may help evaluate emphysema, alternative diagnoses, or complications, and ECG may be used when cardiac disease is suspected.
Management is challenging because many ACO patients have been excluded from major asthma and COPD drug trials, leaving limited high-quality evidence to guide therapy. A central reason to identify ACO is the potential for different responses to inhaled corticosteroids (ICS), particularly in patients with eosinophilic inflammation. Nonpharmacologic management includes smoking cessation, avoidance of triggers, inhaler technique education, and pulmonary rehabilitation; vaccination and oxygen supplementation may also be appropriate depending on severity. Pharmacologic treatment is generally symptom-directed, with bronchodilators used for dynamic obstruction and hyperinflation, while ICS is often included when asthma-like features or eosinophilia are present. Current guidance discourages using LABA and/or LAMA without ICS when asthma features are present, due to safety concerns and the need to treat underlying airway inflammation. Combination ICS/LABA therapy has evidence of benefit in some ACO populations, and observational data suggest improved outcomes compared with bronchodilator therapy alone in patients with overlapping asthma and COPD histories. Selected patients with allergic features may respond to biologic therapy such as omalizumab, with some observational data showing improvements similar to those seen in asthma without ACO.
Long-term care emphasizes earlier recognition in smokers or former smokers with partially reversible obstruction and progressive exercise intolerance, along with comprehensive management of comorbidities that worsen respiratory symptoms—such as GERD, chronic aspiration, vocal cord dysfunction, and nasal/sinus disease. Cardiovascular evaluation is especially important given higher cardiovascular risk in this population. Referral to a specialist is appropriate when symptoms or exacerbations persist, diagnosis remains uncertain, atypical features appear (e.g., hemoptysis, weight loss, fevers, night sweats, suspected bronchiectasis), comorbidities complicate management, or there is poor response to standard therapy. Prevention strategies focus on smoking cessation and minimizing exposure to triggers and irritants.
Asthma–COPD overlap (ACO) is a recognized clinical entity describing patients—often current or former smokers—who have chronic obstructive pulmonary disease (COPD) but also share important inflammatory and clinical features of asthma. Compared with COPD alone, ACO tends to be a more severe phenotype, with greater symptom burden and worse overall outcomes. ACO may be described as asthma with partially reversible airflow obstruction (with or without emphysema or reduced diffusing capacity) or as COPD with emphysema accompanied by reversible or partially reversible airflow obstruction (with or without allergies or reduced diffusing capacity). Because patients present with different patterns—such as COPD with eosinophilia, severe asthma in smokers, neutrophilic-predominant asthma, or asthma that has become largely irreversible due to airway remodeling—the term “ACO” is preferred over the older “ACOS” to reflect this heterogeneity.
ACO is discussed using overlapping ICD-10 codes including asthma (J45), chronic bronchitis (J42), emphysema (J43), COPD (J44, J44.9), and other overlap syndromes (M35.1). Epidemiologically, COPD prevalence varies across regions, and asthma affects tens of millions of people in the U.S. Bronchial hyperresponsiveness is common in COPD and bronchodilator reversibility can also be seen in COPD, which complicates diagnosis. Across studies, patients identified as having ACO generally demonstrate worse lung function, more respiratory symptoms, more frequent exacerbations, higher health-care utilization, and lower quality of life than those with asthma or COPD alone. Reported prevalence of ACO among adults with obstructive airway disease is commonly estimated around 15%–25%, with higher prevalence in older adults and in those with more severe disease. Some data suggest mortality in ACO is similar to COPD and worse than asthma alone.
Risk factors for ACO include cigarette smoking and atopy, along with older age, allergies, higher BMI, childhood asthma history, and respiratory infections such as rhinovirus or influenza. While no definitive genetic basis is established, some research has identified variants (including in GPR65) associated with ACO in certain populations. Clinically, patients may have wheezing, dyspnea, chest tightness, chronic cough (often productive), reduced exercise tolerance, recurrent respiratory infections, and episodic symptoms triggered by allergens, odors, or temperature changes. Physical examination can be normal or may show wheezing or rhonchi; in more severe cases, decreased air entry and accessory muscle use may appear, and signs such as the Hoover sign can suggest hyperinflation.
Diagnosis is considered when a patient demonstrates a mixed pattern of asthma-like and COPD-like features. A practical approach relies on objective confirmation of persistent airflow obstruction plus evidence of reversibility or airway hyperresponsiveness. Patients should have a reduced post-bronchodilator FEV₁/FVC (below the lower limit of normal or <0.7), along with either bronchodilator reversibility (increase in fev₁ or fvc by ≥200 ml and ≥12%) airway hyperresponsiveness demonstrated a positive methacholine challenge. because can occur copd symptoms overlap substantially, aco remains clinical diagnosis supported the pattern of symptoms, smoking history, asthma />llergy features, and pulmonary function testing rather than a single definitive test.
The differential diagnosis includes asthma, COPD, central airway obstruction, bronchiectasis, heart failure, and obliterative bronchiolitis. Workup typically includes spirometry with bronchodilator testing, and sometimes airway provocation testing. Additional supportive testing may include ABG (especially in severe disease), CBC with eosinophils, total IgE, sputum assessment, allergy testing, and peak flow monitoring. Imaging with chest x-ray or chest CT may help evaluate emphysema, alternative diagnoses, or complications, and ECG may be used when cardiac disease is suspected.
Management is challenging because many ACO patients have been excluded from major asthma and COPD drug trials, leaving limited high-quality evidence to guide therapy. A central reason to identify ACO is the potential for different responses to inhaled corticosteroids (ICS), particularly in patients with eosinophilic inflammation. Nonpharmacologic management includes smoking cessation, avoidance of triggers, inhaler technique education, and pulmonary rehabilitation; vaccination and oxygen supplementation may also be appropriate depending on severity. Pharmacologic treatment is generally symptom-directed, with bronchodilators used for dynamic obstruction and hyperinflation, while ICS is often included when asthma-like features or eosinophilia are present. Current guidance discourages using LABA and/or LAMA without ICS when asthma features are present, due to safety concerns and the need to treat underlying airway inflammation. Combination ICS/LABA therapy has evidence of benefit in some ACO populations, and observational data suggest improved outcomes compared with bronchodilator therapy alone in patients with overlapping asthma and COPD histories. Selected patients with allergic features may respond to biologic therapy such as omalizumab, with some observational data showing improvements similar to those seen in asthma without ACO.
Long-term care emphasizes earlier recognition in smokers or former smokers with partially reversible obstruction and progressive exercise intolerance, along with comprehensive management of comorbidities that worsen respiratory symptoms—such as GERD, chronic aspiration, vocal cord dysfunction, and nasal/sinus disease. Cardiovascular evaluation is especially important given higher cardiovascular risk in this population. Referral to a specialist is appropriate when symptoms or exacerbations persist, diagnosis remains uncertain, atypical features appear (e.g., hemoptysis, weight loss, fevers, night sweats, suspected bronchiectasis), comorbidities complicate management, or there is poor response to standard therapy. Prevention strategies focus on smoking cessation and minimizing exposure to triggers and irritants.
- Published on
KembaraXtra-Medicine – Astrocytoma
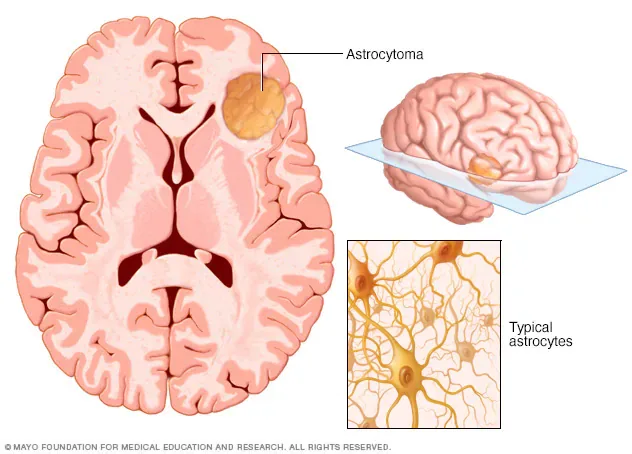
Astrocytomas are neuroepithelial tumors that arise from astrocytes, a type of glial precursor cell in the central nervous system. They are classified by the World Health Organization (WHO) into four grades based on histopathologic features, which provide important prognostic information. Grade I tumors are pilocytic astrocytomas and are generally benign. Grade II tumors are diffuse astrocytomas and are considered low grade. Grade III tumors are anaplastic astrocytomas, and Grade IV tumors are glioblastomas. Grades III and IV are classified as high-grade or malignant astrocytomas.
Astrocytomas account for approximately 10% of primary CNS tumors in the United States. In 2022, there were an estimated 25,050 new cases and 18,280 deaths related to primary CNS tumors, with an overall incidence of 6.4 cases per 100,000 persons per year. About half of malignant CNS tumors are glioblastomas. The clinical presentation of astrocytoma depends largely on tumor location and growth rate. Patients may present with headaches related to mass effect, new-onset partial or generalized seizures, nausea, vomiting, fatigue, and weakness. Focal neurologic deficits such as cranial nerve palsies, hemiplegia, ataxia, visual impairment, or language disturbances may occur. Cognitive or personality changes and, rarely, papilledema can also be seen.
The exact cause of astrocytomas remains unclear, although certain risk factors have been identified. Exposure to ionizing radiation is a known risk factor, and higher incidence has been observed in farmers and petrochemical workers. Several hereditary conditions are associated with increased risk, including neurofibromatosis type 1, Lynch syndrome, and Li-Fraumeni syndrome. At the molecular level, alterations in tumor suppressor genes such as TP53 play a significant role in astrocytoma development. Mutations in the IDH1 gene are common in grade II and III astrocytomas and are associated with a better prognosis compared with IDH-wild-type tumors. Molecular classification using TERT promoter mutations, IDH mutations, and 1p/19q codeletion further refines prognosis and treatment planning.
Diagnosis of astrocytoma is initially suspected based on clinical presentation and neuroimaging, but definitive diagnosis and grading require histopathologic confirmation. The differential diagnosis is broad and includes other causes of headaches, seizures, altered mental status, and focal neurologic deficits. Contrast-enhanced MRI is the imaging modality of choice because it provides detailed anatomic and pathologic information. CT scanning is reserved for patients unable to undergo MRI. Tissue diagnosis is obtained through surgical resection or stereotactic biopsy, particularly for deep-seated, multifocal, or unresectable tumors. Surgical goals include maximal safe tumor removal, reduction of mass effect and intracranial pressure, and procurement of tissue for diagnosis.
Management of astrocytoma is stage-specific and multidisciplinary. Corticosteroids such as dexamethasone are commonly used perioperatively to reduce cerebral edema. Maximal safe surgical resection is the cornerstone of treatment for all grades when feasible. Grade I astrocytomas are often cured with complete resection, while unresectable tumors may be observed until progression. Grade II astrocytomas benefit from surgery followed by adjuvant radiotherapy and chemotherapy, particularly when residual disease remains. Grade III anaplastic astrocytomas are treated with surgery followed by combined radiotherapy and chemotherapy, most commonly temozolomide. Grade IV glioblastomas require aggressive multimodal therapy, including maximal resection, concurrent chemoradiation with temozolomide, and adjuvant chemotherapy. Tumor-treating fields have shown additional survival benefit and are FDA-approved in selected patients.
Recurrent disease management depends on tumor grade and prior therapy. Options include repeat surgical resection, radiotherapy, chemotherapy, targeted therapies, and experimental approaches such as immunotherapy, tumor vaccines, and CAR-T cell therapy. Prognosis varies by grade, with excellent outcomes for grade I tumors, median survival of approximately 7.5 years for grade II, about 5 years for grade III, and roughly 14 months for glioblastoma. IDH-mutated tumors and those with favorable molecular features have improved survival.
Care of patients with astrocytoma requires coordinated management by neurosurgery, radiation oncology, and neuro-oncology teams to guide diagnosis, treatment, and long-term follow-up.
Astrocytomas are neuroepithelial tumors that arise from astrocytes, a type of glial precursor cell in the central nervous system. They are classified by the World Health Organization (WHO) into four grades based on histopathologic features, which provide important prognostic information. Grade I tumors are pilocytic astrocytomas and are generally benign. Grade II tumors are diffuse astrocytomas and are considered low grade. Grade III tumors are anaplastic astrocytomas, and Grade IV tumors are glioblastomas. Grades III and IV are classified as high-grade or malignant astrocytomas.
Astrocytomas account for approximately 10% of primary CNS tumors in the United States. In 2022, there were an estimated 25,050 new cases and 18,280 deaths related to primary CNS tumors, with an overall incidence of 6.4 cases per 100,000 persons per year. About half of malignant CNS tumors are glioblastomas. The clinical presentation of astrocytoma depends largely on tumor location and growth rate. Patients may present with headaches related to mass effect, new-onset partial or generalized seizures, nausea, vomiting, fatigue, and weakness. Focal neurologic deficits such as cranial nerve palsies, hemiplegia, ataxia, visual impairment, or language disturbances may occur. Cognitive or personality changes and, rarely, papilledema can also be seen.
The exact cause of astrocytomas remains unclear, although certain risk factors have been identified. Exposure to ionizing radiation is a known risk factor, and higher incidence has been observed in farmers and petrochemical workers. Several hereditary conditions are associated with increased risk, including neurofibromatosis type 1, Lynch syndrome, and Li-Fraumeni syndrome. At the molecular level, alterations in tumor suppressor genes such as TP53 play a significant role in astrocytoma development. Mutations in the IDH1 gene are common in grade II and III astrocytomas and are associated with a better prognosis compared with IDH-wild-type tumors. Molecular classification using TERT promoter mutations, IDH mutations, and 1p/19q codeletion further refines prognosis and treatment planning.
Diagnosis of astrocytoma is initially suspected based on clinical presentation and neuroimaging, but definitive diagnosis and grading require histopathologic confirmation. The differential diagnosis is broad and includes other causes of headaches, seizures, altered mental status, and focal neurologic deficits. Contrast-enhanced MRI is the imaging modality of choice because it provides detailed anatomic and pathologic information. CT scanning is reserved for patients unable to undergo MRI. Tissue diagnosis is obtained through surgical resection or stereotactic biopsy, particularly for deep-seated, multifocal, or unresectable tumors. Surgical goals include maximal safe tumor removal, reduction of mass effect and intracranial pressure, and procurement of tissue for diagnosis.
Management of astrocytoma is stage-specific and multidisciplinary. Corticosteroids such as dexamethasone are commonly used perioperatively to reduce cerebral edema. Maximal safe surgical resection is the cornerstone of treatment for all grades when feasible. Grade I astrocytomas are often cured with complete resection, while unresectable tumors may be observed until progression. Grade II astrocytomas benefit from surgery followed by adjuvant radiotherapy and chemotherapy, particularly when residual disease remains. Grade III anaplastic astrocytomas are treated with surgery followed by combined radiotherapy and chemotherapy, most commonly temozolomide. Grade IV glioblastomas require aggressive multimodal therapy, including maximal resection, concurrent chemoradiation with temozolomide, and adjuvant chemotherapy. Tumor-treating fields have shown additional survival benefit and are FDA-approved in selected patients.
Recurrent disease management depends on tumor grade and prior therapy. Options include repeat surgical resection, radiotherapy, chemotherapy, targeted therapies, and experimental approaches such as immunotherapy, tumor vaccines, and CAR-T cell therapy. Prognosis varies by grade, with excellent outcomes for grade I tumors, median survival of approximately 7.5 years for grade II, about 5 years for grade III, and roughly 14 months for glioblastoma. IDH-mutated tumors and those with favorable molecular features have improved survival.
Care of patients with astrocytoma requires coordinated management by neurosurgery, radiation oncology, and neuro-oncology teams to guide diagnosis, treatment, and long-term follow-up.
- Published on
KembaraXtra-Medicine- Acute Glomerulonephritis
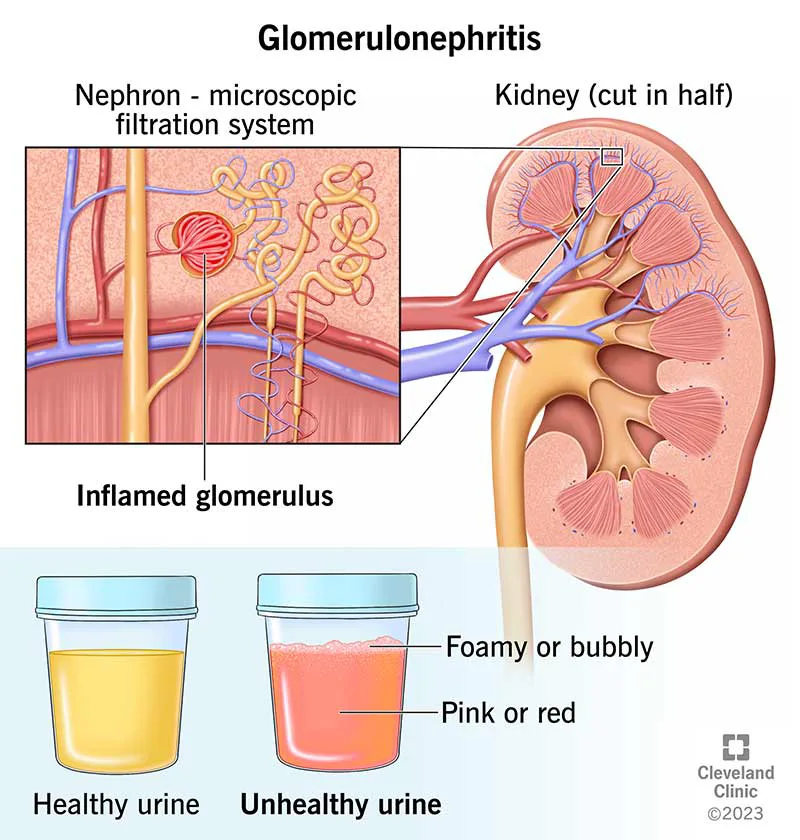
Acute glomerulonephritis (GN) is inflammation of the kidney glomerulus—an intricate capillary network—most often driven by an autoimmune process that causes deposition of immunoreactants and recruitment of inflammatory cells. Clinically, it typically presents with hematuria and proteinuria along with new or worsening hypertension. Without appropriate treatment, ongoing glomerular inflammation can progress to chronic kidney disease.
Acute GN is also referred to as acute nephritic syndrome (or acute GN). ICD-10-CM coding falls under the acute nephritic syndrome category (N00.0–N00.9), with codes reflecting morphologic patterns such as focal/segmental lesions, diffuse membranous disease, mesangial proliferative patterns, endocapillary proliferative GN, mesangiocapillary/MPGN patterns, dense deposit disease, crescentic GN, and unspecified morphologic changes.
Epidemiologically, the incidence of primary GN varies by subtype, with reported estimates ranging from about 0.2 per 100,000 patient-years for membranoproliferative GN to about 2.5 per 100,000 patient-years for IgA nephropathy. IgA nephropathy is the most common form worldwide. Glomerulonephritis contributes substantially to global kidney failure burden, accounting for a significant proportion of end-stage kidney disease. GN occurs in both children and adults.
Patients commonly present with acute-onset hypertension, dark “tea-colored” urine from gross hematuria, edema (periorbital, peripheral, or pulmonary), and fatigue. Certain systemic clues suggest an underlying cause: concurrent pulmonary hemorrhage with rapidly progressive renal decline suggests crescentic GN (often ANCA-associated vasculitis or anti-GBM disease). Joint pain, oral ulcers, and malar rash point toward systemic lupus erythematosus (SLE). Palpable purpura can be seen in systemic vasculitides such as IgA vasculitis, ANCA-associated vasculitis, SLE, or cryoglobulinemia. A recent history of pharyngitis, cellulitis, or endocarditis preceding urinary abnormalities raises suspicion for infection-related GN. Hepatitis C can cause membranoproliferative GN with or without cryoglobulinemia, and “synpharyngitic” gross hematuria (hematuria coinciding with an upper respiratory infection) is a classic pattern in IgA nephropathy.
Etiologically, acute GN can be kidney-limited or part of a broader systemic disorder. Understanding of mechanisms has expanded considerably. IgA nephropathy is described as a multi-hit process involving under–O-galactosylated IgA1, formation of O-glycan–specific antibodies, and deposition of IgA1-containing immune complexes in glomeruli. Hepatitis C can drive chronic immune stimulation with cryoglobulin formation and deposition. Poststreptococcal GN is now included within “infection-related GN,” which also includes IgA-dominant staphylococcal-associated GN. ANCA-associated vasculitis is linked to antibodies against myeloperoxidase and proteinase 3, though direct causation remains complex. Paraprotein-related glomerular injury can occur even without overt hematologic malignancy; when kidney injury is driven by a monoclonal immunoglobulin in that setting, it is termed monoclonal gammopathy of renal significance. C3 glomerulopathy (C3 GN and dense deposit disease) reflects dysregulation of the alternative complement pathway, and complement activation is increasingly recognized across multiple GN entities. Thrombotic microangiopathy in native kidneys results from endothelial injury triggered by drugs, autoimmune disorders, infection, or genetic causes.
The differential diagnosis for hematuria and/or proteinuria is broad and includes urinary tract infection, nephrolithiasis, urothelial malignancy, polycystic kidney disease, acute interstitial nephritis, acute tubular necrosis, nephrotic syndrome, hereditary nephritis (e.g., Alport syndrome or thin basement membrane nephropathy), and diabetic nephropathy. These should be considered alongside GN based on the clinical picture and laboratory findings.
Workup begins with laboratory testing. Urinalysis typically shows albuminuria (the predominant protein) and hematuria with dysmorphic red blood cells and/or red cell casts. Serum blood urea nitrogen and creatinine assess kidney function. Quantification of proteinuria can be done by a 24-hour urine collection or, more commonly in practice, a spot urine protein-to-creatinine ratio; proteinuria in acute GN often ranges from about 500 mg/day to 3 g/day, though nephrotic-range proteinuria can occur. If infection-related GN is suspected, streptococcal testing (e.g., Streptozyme) and antistreptolysin O (ASO) titers can be helpful; ASO typically peaks around 3 to 5 weeks and does not correlate with disease severity or prognosis. Broader serologic evaluation is guided by suspicion for systemic disease and commonly includes ANA, anti–double-stranded DNA, complement levels (C3/C4), hepatitis B and C serologies, HIV testing, ANCA (MPO and PR3), anti-GBM antibodies, cryoglobulins, rheumatoid factor, serum and urine protein electrophoresis with immunofixation, and serum free light chains. In thrombotic microangiopathy, hematocrit and platelets may be decreased, though renal-limited cases can have normal counts. Febrile patients should have blood cultures obtained.
Imaging supports evaluation and identifies complications or alternative diagnoses. A chest radiograph is important when pulmonary symptoms or opacities are present, because diffuse alveolar hemorrhage can accompany ANCA-associated vasculitis or anti-GBM (Goodpasture disease). Renal ultrasound helps exclude structural causes of hematuria/proteinuria and provides kidney size; a sagittal length below about 9 cm suggests chronic scarring and limited reversibility. Echocardiography is indicated when there is a new murmur or positive blood cultures to assess for endocarditis and to evaluate for pericardial effusion. Definitive diagnosis and classification often require kidney biopsy with light microscopy, immunofluorescence, and electron microscopy, and biopsy of other affected organs may be considered when systemic vasculitis is suspected.
Treatment is highly dependent on the specific GN subtype and severity and should be directed with urgent nephrology involvement, especially when there is azotemia, hyperkalemia, or metabolic acidosis. Supportive nonpharmacologic measures include a low-sodium diet (around 2 g/day) for edema or hypertension and avoiding high-potassium foods when hyperkalemia is present. Acute management frequently includes diuretics for edema or hypertension, correction of electrolyte abnormalities and metabolic acidosis, and hemodialysis for diuretic-resistant volume overload, refractory hyperkalemia, uremic symptoms, or severe acidosis. Plasma exchange for antibody removal in rapidly progressive GN or diffuse alveolar hemorrhage is no longer a routine evidence-based standard for all such cases, but it may be considered selectively depending on the clinical scenario and underlying diagnosis.
Chronic management focuses on monitoring for relapse or progression and mitigating long-term complications. Many GN types may follow a relapsing-remitting course, so periodic monitoring of blood pressure, urinalysis, renal function (creatinine/BUN), serum albumin, and urine protein-to-creatinine ratio is important. ACE inhibitors or ARBs are commonly used to reduce proteinuria when not contraindicated. Lipid management with statins (and fibrates when indicated) is important, particularly when nephrotic-range proteinuria or dyslipidemia is present. Patients receiving immunosuppression require monitoring for infection, cytopenias, bone loss, gastrointestinal ulcers, hypertension, and malignancy risk, along with routine preventive care and vaccination per CDC guidance for chronic kidney disease and immunocompromised states; live vaccines are contraindicated during active immunosuppressive therapy.
Prognosis correlates strongly with initial kidney function (serum creatinine/eGFR) and the degree of chronic damage and fibrosis on kidney biopsy. Outcomes are generally worse in patients with heavy albuminuria/proteinuria, low GFR at presentation, severe hypertension, and biopsy-proven crescentic GN with a high proportion of crescents. Nephrology consultation is recommended for all suspected GN, and urgent consultation is warranted when hyperkalemia, acidosis, or significant azotemia is present.
Acute glomerulonephritis (GN) is inflammation of the kidney glomerulus—an intricate capillary network—most often driven by an autoimmune process that causes deposition of immunoreactants and recruitment of inflammatory cells. Clinically, it typically presents with hematuria and proteinuria along with new or worsening hypertension. Without appropriate treatment, ongoing glomerular inflammation can progress to chronic kidney disease.
Acute GN is also referred to as acute nephritic syndrome (or acute GN). ICD-10-CM coding falls under the acute nephritic syndrome category (N00.0–N00.9), with codes reflecting morphologic patterns such as focal/segmental lesions, diffuse membranous disease, mesangial proliferative patterns, endocapillary proliferative GN, mesangiocapillary/MPGN patterns, dense deposit disease, crescentic GN, and unspecified morphologic changes.
Epidemiologically, the incidence of primary GN varies by subtype, with reported estimates ranging from about 0.2 per 100,000 patient-years for membranoproliferative GN to about 2.5 per 100,000 patient-years for IgA nephropathy. IgA nephropathy is the most common form worldwide. Glomerulonephritis contributes substantially to global kidney failure burden, accounting for a significant proportion of end-stage kidney disease. GN occurs in both children and adults.
Patients commonly present with acute-onset hypertension, dark “tea-colored” urine from gross hematuria, edema (periorbital, peripheral, or pulmonary), and fatigue. Certain systemic clues suggest an underlying cause: concurrent pulmonary hemorrhage with rapidly progressive renal decline suggests crescentic GN (often ANCA-associated vasculitis or anti-GBM disease). Joint pain, oral ulcers, and malar rash point toward systemic lupus erythematosus (SLE). Palpable purpura can be seen in systemic vasculitides such as IgA vasculitis, ANCA-associated vasculitis, SLE, or cryoglobulinemia. A recent history of pharyngitis, cellulitis, or endocarditis preceding urinary abnormalities raises suspicion for infection-related GN. Hepatitis C can cause membranoproliferative GN with or without cryoglobulinemia, and “synpharyngitic” gross hematuria (hematuria coinciding with an upper respiratory infection) is a classic pattern in IgA nephropathy.
Etiologically, acute GN can be kidney-limited or part of a broader systemic disorder. Understanding of mechanisms has expanded considerably. IgA nephropathy is described as a multi-hit process involving under–O-galactosylated IgA1, formation of O-glycan–specific antibodies, and deposition of IgA1-containing immune complexes in glomeruli. Hepatitis C can drive chronic immune stimulation with cryoglobulin formation and deposition. Poststreptococcal GN is now included within “infection-related GN,” which also includes IgA-dominant staphylococcal-associated GN. ANCA-associated vasculitis is linked to antibodies against myeloperoxidase and proteinase 3, though direct causation remains complex. Paraprotein-related glomerular injury can occur even without overt hematologic malignancy; when kidney injury is driven by a monoclonal immunoglobulin in that setting, it is termed monoclonal gammopathy of renal significance. C3 glomerulopathy (C3 GN and dense deposit disease) reflects dysregulation of the alternative complement pathway, and complement activation is increasingly recognized across multiple GN entities. Thrombotic microangiopathy in native kidneys results from endothelial injury triggered by drugs, autoimmune disorders, infection, or genetic causes.
The differential diagnosis for hematuria and/or proteinuria is broad and includes urinary tract infection, nephrolithiasis, urothelial malignancy, polycystic kidney disease, acute interstitial nephritis, acute tubular necrosis, nephrotic syndrome, hereditary nephritis (e.g., Alport syndrome or thin basement membrane nephropathy), and diabetic nephropathy. These should be considered alongside GN based on the clinical picture and laboratory findings.
Workup begins with laboratory testing. Urinalysis typically shows albuminuria (the predominant protein) and hematuria with dysmorphic red blood cells and/or red cell casts. Serum blood urea nitrogen and creatinine assess kidney function. Quantification of proteinuria can be done by a 24-hour urine collection or, more commonly in practice, a spot urine protein-to-creatinine ratio; proteinuria in acute GN often ranges from about 500 mg/day to 3 g/day, though nephrotic-range proteinuria can occur. If infection-related GN is suspected, streptococcal testing (e.g., Streptozyme) and antistreptolysin O (ASO) titers can be helpful; ASO typically peaks around 3 to 5 weeks and does not correlate with disease severity or prognosis. Broader serologic evaluation is guided by suspicion for systemic disease and commonly includes ANA, anti–double-stranded DNA, complement levels (C3/C4), hepatitis B and C serologies, HIV testing, ANCA (MPO and PR3), anti-GBM antibodies, cryoglobulins, rheumatoid factor, serum and urine protein electrophoresis with immunofixation, and serum free light chains. In thrombotic microangiopathy, hematocrit and platelets may be decreased, though renal-limited cases can have normal counts. Febrile patients should have blood cultures obtained.
Imaging supports evaluation and identifies complications or alternative diagnoses. A chest radiograph is important when pulmonary symptoms or opacities are present, because diffuse alveolar hemorrhage can accompany ANCA-associated vasculitis or anti-GBM (Goodpasture disease). Renal ultrasound helps exclude structural causes of hematuria/proteinuria and provides kidney size; a sagittal length below about 9 cm suggests chronic scarring and limited reversibility. Echocardiography is indicated when there is a new murmur or positive blood cultures to assess for endocarditis and to evaluate for pericardial effusion. Definitive diagnosis and classification often require kidney biopsy with light microscopy, immunofluorescence, and electron microscopy, and biopsy of other affected organs may be considered when systemic vasculitis is suspected.
Treatment is highly dependent on the specific GN subtype and severity and should be directed with urgent nephrology involvement, especially when there is azotemia, hyperkalemia, or metabolic acidosis. Supportive nonpharmacologic measures include a low-sodium diet (around 2 g/day) for edema or hypertension and avoiding high-potassium foods when hyperkalemia is present. Acute management frequently includes diuretics for edema or hypertension, correction of electrolyte abnormalities and metabolic acidosis, and hemodialysis for diuretic-resistant volume overload, refractory hyperkalemia, uremic symptoms, or severe acidosis. Plasma exchange for antibody removal in rapidly progressive GN or diffuse alveolar hemorrhage is no longer a routine evidence-based standard for all such cases, but it may be considered selectively depending on the clinical scenario and underlying diagnosis.
Chronic management focuses on monitoring for relapse or progression and mitigating long-term complications. Many GN types may follow a relapsing-remitting course, so periodic monitoring of blood pressure, urinalysis, renal function (creatinine/BUN), serum albumin, and urine protein-to-creatinine ratio is important. ACE inhibitors or ARBs are commonly used to reduce proteinuria when not contraindicated. Lipid management with statins (and fibrates when indicated) is important, particularly when nephrotic-range proteinuria or dyslipidemia is present. Patients receiving immunosuppression require monitoring for infection, cytopenias, bone loss, gastrointestinal ulcers, hypertension, and malignancy risk, along with routine preventive care and vaccination per CDC guidance for chronic kidney disease and immunocompromised states; live vaccines are contraindicated during active immunosuppressive therapy.
Prognosis correlates strongly with initial kidney function (serum creatinine/eGFR) and the degree of chronic damage and fibrosis on kidney biopsy. Outcomes are generally worse in patients with heavy albuminuria/proteinuria, low GFR at presentation, severe hypertension, and biopsy-proven crescentic GN with a high proportion of crescents. Nephrology consultation is recommended for all suspected GN, and urgent consultation is warranted when hyperkalemia, acidosis, or significant azotemia is present.
- Published on
KembaraXtra- Medicine – Acute Myeloid Leukemia (AML)
Acute myeloid leukemia (AML) is a malignancy of hematopoietic progenitor cells that normally mature into granulocytes. More broadly, AML sits within the acute nonlymphocytic leukemia (ANLL) spectrum, which includes leukemias arising from myeloid stem cells and precursors of granulocytes, monocytes, erythrocytes, and megakaryocytes, distinguishing them from leukemias of lymphocytic origin. AML is characterized by maturation failure of myeloid progenitors, accumulation of immature blasts, and bone marrow failure leading to varying degrees of neutropenia, thrombocytopenia, and anemia. Acute promyelocytic leukemia (APL/APML) is a distinct AML syndrome with very different treatment implications and must be recognized urgently due to its high risk of catastrophic bleeding.
Synonyms include acute nonlymphocytic leukemia (ANLL), acute myelogenous leukemia, and AML. ICD-10-CM codes include C92.00–C92.02 (acute myeloblastic leukemia categories), C92.60–C92.62 (AML with 11q23 abnormality categories), C92.90–C92.92 (myeloid leukemia unspecified categories), C92.A0–C92.A2 (AML with multilineage dysplasia categories), and C92.Z0–C92.Z2 (other myeloid leukemia categories).
AML incidence rises with age. In adults aged 20–55 years, incidence is approximately 1–3 per 100,000 persons/year, increasing to 11–20 per 100,000 persons/year in those aged 65–80 years, representing about a tenfold increase. The median age at diagnosis is ~68 years (SEER 2015–2019). Overall annual incidence is about 4 per 100,000 persons/year. Males are affected slightly more than females, and individuals of European ancestry slightly more than those of African ancestry.
Clinical presentation is commonly due to bone marrow failure and/or hyperleukocytosis. Cytopenia-related findings include bleeding from thrombocytopenia, fatigue and shortness of breath from anemia, and infections from neutropenia. Systemic symptoms may include fatigue, fever (usually infectious, less commonly tumor-related), and sometimes bone pain (noted as more common in ALL than AML). Physical examination often reflects cytopenias (e.g., pallor, bruising/petechiae), while lymphadenopathy and hepatosplenomegaly are rare, and the exam may otherwise be normal.
A critical “don’t miss” scenario is hyperleukocytic leukemia, typically with WBC >100,000/µL, which can cause leukostasis and ischemic complications. These include retinal hemorrhage and visual symptoms (blurred vision, diplopia), intracranial bleeding or neurologic deficits (headache, confusion, delirium, paralysis), respiratory compromise (dyspnea, tachypnea, hypoxia from pulmonary involvement), and organ ischemia (stroke, myocardial infarction, priapism). Another high-risk complication is disseminated intravascular coagulation (DIC), especially prominent in APL, though DIC can occur in other acute leukemias (notably acute monocytic leukemia). AML may rarely present as extramedullary disease such as leukemia cutis, blastic plasmacytoid dendritic cell neoplasm (BPDCN), or granulocytic sarcoma. Gingival hypertrophy and tissue/organ involvement are more typical of monocytic leukemia.
Etiology and risk contributors include environmental exposures (with benzene best documented), organic solvents, cigarette smoking, and obesity (best documented in women). Hereditary predisposition syndromes include bone marrow failure syndromes (Fanconi anemia, Bloom syndrome, Shwachman-Diamond syndrome, Diamond-Blackfan anemia), genetic disorders (e.g., Down syndrome), and familial AML syndromes (e.g., DDX41, RUNX1, GATA2 mutations), among others. Therapy-related AML (t-AML) accounts for roughly 10%–20% of cases and classically occurs after exposure to alkylating agents (latency 5–7 years, often associated with chromosome 5/7 abnormalities and/or TP53) or topoisomerase II inhibitors (latency 1–3 years, often associated with 11q23/KMT2A (MLL) rearrangements). Radiation exposure can contribute, and AML may also arise after antecedent hematologic disorders (myelodysplasia, myeloproliferative disorders, aplastic anemia). Clonal hematopoiesis of indeterminate potential (CHIP) is increasingly recognized as a precursor state that may predispose to AML years later.
The differential diagnosis includes other disorders with circulating blasts or blast-like cells: AML/ALL, myelodysplasia (which can show up to 20% circulating blasts; ≥20% blasts meets AML criteria), primary myelofibrosis, chronic myeloid leukemia, blastoid mantle cell lymphoma, prolymphocytic leukemia, BPDCN, and reactive atypical lymphocytes in EBV/CMV infections that may appear blast-like.
Diagnosis requires combined assessment of morphology, immunophenotyping, cytogenetics, and molecular testing. Initial evaluation includes CBC and peripheral smear, recognizing that morphology can suggest lineage but flow cytometry and/or cytochemistry are needed for confirmation. Auer rods support myeloid lineage. LDH is commonly elevated. Baseline labs assess organ function (creatinine, liver enzymes) and screen for spontaneous tumor lysis syndrome (uric acid, potassium, phosphate, calcium). Vitamin B12 and folate can be low due to rapid turnover and may require replacement. Coagulation studies (PT, aPTT, fibrinogen) are essential to evaluate for DIC, which is always present in APL and can occur in other acute leukemias. HLA typing supports transplant planning and platelet support.
Cytochemical stains may be rapidly available: myeloperoxidase (MPO) positivity supports myeloid origin and can be obtained quickly, while nonspecific esterase supports monocytic differentiation. Flow cytometry on blood and/or marrow typically confirms lineage and maturation stage and uses marker patterns such as precursor markers (CD34, CD117, CD13, CD133, HLA-DR), granulocytic markers (CD65, cytoplasmic MPO), monocytic markers (CD14, CD36, CD64), megakaryocytic markers (CD41, CD61), and erythroid markers (CD235, CD36). Cytogenetic studies (karyotype) are ideally performed on bone marrow but can be done on peripheral blood; FISH is commonly used as an adjunct. Next-generation sequencing / PCR panels detect prognostic and targetable mutations—commonly FLT3, NPM1, IDH1/IDH2, and CEBPA—and broader panels are increasingly used because of evolving prognostic frameworks and targeted therapy availability.
A formal AML diagnosis is established when blasts are ≥20% in the marrow or peripheral blood, unless AML-defining recurrent cytogenetic abnormalities are present—especially t(8;21), inv(16)/t(16;16), or t(15;17)—in which case AML can be diagnosed regardless of blast percentage. Risk stratification commonly follows genetic groupings: favorable-risk disease includes core-binding factor AML (e.g., t(8;21), inv(16)/t(16;16)) and certain normal-karyotype molecular profiles (e.g., mutated NPM1 without FLT3-ITD, or favorable CEBPA patterns), intermediate-risk includes several mixed molecular/cytogenetic patterns, and adverse-risk includes lesions such as inv(3)/t(3;3), t(6;9), KMT2A/MLL rearrangements, t(9;22), and RUNX1/ASXL1/TP53 mutations, as well as -5/del(5q), -7, abnl(17p), and complex karyotype.
Treatment is typically organized into urgent stabilization plus disease-directed therapy. Immediate priorities include correction of life-threatening complications (infection/sepsis risk in neutropenia, bleeding/DIC especially in APL, tumor lysis syndrome, and leukostasis/hyperleukocytosis). AML therapy generally includes induction to achieve remission (commonly “7+3”: daunorubicin for 3 days plus cytarabine continuous infusion for 7 days in many fit patients), followed by consolidation to prevent relapse (often intermediate/high-dose cytarabine in favorable-risk disease, and consideration of allogeneic stem cell transplant in intermediate/unfavorable-risk disease when appropriate). Hyperleukocytosis may require rapid cytoreduction (e.g., hydroxyurea and/or chemotherapy) and sometimes leukapheresis depending on clinical context. Tumor lysis syndrome prevention and management relies on vigorous hydration, close electrolyte monitoring, and uric-acid–lowering strategies as indicated.
A major “don’t miss” subtype is acute promyelocytic leukemia (APL/APML), associated with t(15;17) PML-RARA, which presents with high hemorrhagic risk due to DIC and requires immediate initiation of all-trans retinoic acid (ATRA) when suspected—often before full molecular confirmation—along with aggressive supportive transfusion strategies to maintain fibrinogen and platelet thresholds. Modern APL protocols (ATRA + arsenic trioxide for many risk groups, with intensified approaches in higher-risk cases) achieve very high cure rates, but early death from bleeding remains a key preventable risk.
Pearls and key considerations include that AML can be an emergency depending on presentation; evaluation should be rapid and coordinated with hematology and laboratory expertise. APL is a true medical emergency because early ATRA can rapidly improve coagulopathy and reduce fatal hemorrhage risk.

- Published on
KembaraXtra-Medicine – Acute Mesenteric Ischemia (AMI)
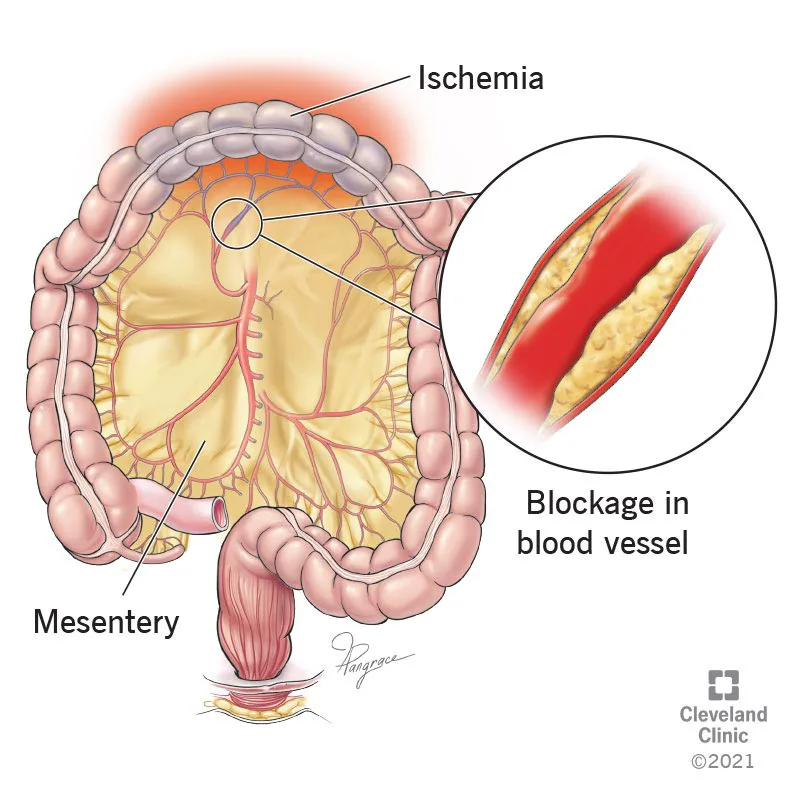
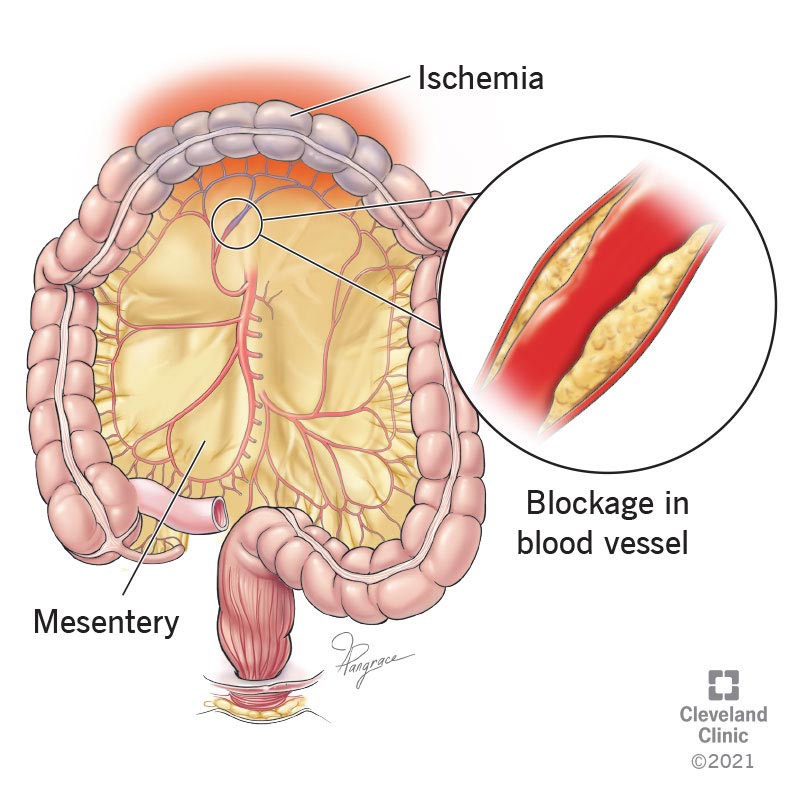
Acute mesenteric ischemia (AMI) is the sudden onset of intestinal hypoperfusion involving all or part of the small bowel due to arterial embolism, arterial thrombosis, mesenteric venous thrombosis, or nonocclusive low-flow/vasospastic states. Reduced blood flow leads to ischemia and secondary inflammatory injury, and—if prolonged—progresses to transmural infarction, necrosis, perforation, sepsis, and death. AMI is also termed acute mesenteric ischemia and is coded as ICD-10-CM K55.0 (acute vascular disorders of intestine).
AMI is uncommon but highly lethal. It accounts for roughly 0.09%–0.2% of emergency admissions and carries a 50%–80% mortality, largely because early disease may have minimal exam findings and diagnosis is frequently delayed. Incidence appears to be increasing, likely from an aging population, improved ICU survival of high-risk patients, increased clinician awareness, and improved imaging access. Arterial embolism and thrombosis are more common in older patients, whereas mesenteric venous thrombosis often affects younger age groups.
Risk factors depend on the AMI subtype. For arterial embolism or thrombosis, key risks include advanced age, atherosclerosis, low cardiac output states (notably atrial fibrillation), severe valvular disease, and intraabdominal malignancy. Mesenteric venous thrombosis is associated with hypercoagulable states (acquired or inherited), portal hypertension, abdominal infection, blunt trauma, pancreatitis, portal malignancy, intraabdominal sepsis, smoking, oral contraceptives, and cirrhosis. Nonocclusive mesenteric ischemia (NOMI) is linked to low-flow states and vasoconstriction, including recent cardiac or aortic surgery, dialysis, hypovolemia, shock, and vasoconstrictive medications and illicit drugs (notably cocaine). AMI can still occur rarely with no identifiable risk factors.
The classic clinical presentation is sudden, severe periumbilical abdominal pain “out of proportion” to the physical examination, often with nausea/vomiting and sometimes diarrhea and/or blood per rectum. About one-third present with the triad of abdominal pain, fever, and hemoccult-positive stool. Early abdominal examination may be deceptively benign—sometimes normal—though mild distention or occult blood may be present; an epigastric bruit can occur. Mesenteric venous thrombosis typically presents with a less abrupt onset than acute arterial occlusion. As ischemia progresses, patients can develop gross distention, absent bowel sounds, sepsis, shock, and peritoneal signs. In older adults, mental status changes may be prominent. Peritonitis strongly suggests irreversible ischemia and bowel necrosis and should trigger immediate surgical action without delay.
AMI is conventionally divided into four etiologic groups with distinct mechanisms and typical patterns. Superior mesenteric artery (SMA) embolism often arises from a cardiac source (left atrium in atrial fibrillation, left ventricle with poor ejection fraction, or valves in endocarditis). Emboli commonly lodge 3–10 cm distal to the SMA origin, often sparing the proximal mid-jejunum. SMA thrombosis usually occurs on a background of chronic atherosclerotic stenosis and can be precipitated by hypotension or low-flow states, producing acute-on-chronic occlusion—often at the SMA orifice—with a larger middle-gut territory at risk. NOMI results from profound splanchnic vasoconstriction in shock/low flow or vasoconstrictor exposure and may involve extensive bowel segments. Mesenteric venous thrombosis causes impaired venous outflow (SMV/IMV/portal system), leading to bowel wall edema, distention, increased resistance, and secondary reduction in arterial inflow. Less common causes include mesenteric artery dissection or inflammation.
Approximate frequencies vary by series, but the provided breakdown includes SMA thrombosis (54%–68%), SMA embolus (26%–32%), NOMI (~10%), mesenteric venous thrombosis (~5%), and focal segmental small-intestine ischemia (~5%). Because outcomes worsen rapidly with time, the most important diagnostic “don’t miss” principle is early suspicion, especially in patients with risk factors and disproportionate pain.
The differential diagnosis is broad early on and should include other causes of acute abdominal pain and peritonitis such as perforated peptic ulcer, pancreatitis, early appendicitis, and other intraabdominal catastrophes. Workup begins with rapid stabilization and parallel diagnostic steps. Laboratory abnormalities classically include leukocytosis, metabolic acidosis, elevated D-dimer, and elevated lactate, but these findings are nonspecific and often late. A key pitfall is relying on lactate early: it may be normal initially and rises more reliably after progression toward necrosis. Serum lactate >2 mmol/L correlates with irreversible transmural necrosis, making it more of a severity marker than an early screening test. D-dimer may help: a normal D-dimer can support ruling out AMI, but an elevated D-dimer is nonspecific. Amylase may be elevated in up to half of cases; phosphate may be elevated in many cases, again typically later. If venous thrombosis is suspected, hypercoagulable evaluation (proteins C/S, antithrombin III, factor V Leiden) may guide long-term therapy but typically does not change acute diagnosis.
CT angiography (CTA) with contrast is the current gold-standard imaging test for suspected AMI because it is fast, widely available, and highly accurate (reported 95%–100% accuracy for visceral ischemic syndromes). CTA can identify arterial occlusion, venous thrombosis, bowel wall changes, and alternate diagnoses, and it may detect embolic sources or other pathology. Plain CT findings are often nonspecific and late; portal venous gas and pneumatosis intestinalis are late findings suggesting gangrene/necrosis and mandate urgent surgery. MRI/MRA can detect proximal SMA/celiac obstruction and may be useful in mesenteric venous thrombosis follow-up, but it is slower and can overestimate stenosis; it is not preferred when CTA is available. Plain abdominal radiographs are frequently normal early and are generally not helpful except for late or indirect signs (ileus, bowel wall thickening, pneumatosis, portal venous gas) or when free air is present (which accelerates the need for surgery). Ultrasound is not recommended if CTA is available because it is time-consuming and limited by bowel gas and body habitus. Conventional angiography is now typically reserved for unclear cases after CT/MRI or when used as part of endovascular intervention planning.
Treatment is time-critical and aims to restore perfusion before infarction while supporting physiology and preventing sepsis. Initial management includes aggressive monitoring and hemodynamic support: crystalloid resuscitation and blood products as needed, correction of acidosis, adequate analgesia (often parenteral opioids), broad-spectrum antibiotics covering gram-negative and anaerobic organisms, and nasogastric decompression. Vasopressors should be used cautiously, only when necessary, to avoid worsening mesenteric hypoperfusion and to reduce risk of abdominal compartment syndrome; when inotropic support is needed, agents such as dobutamine, low-dose dopamine, or milrinone may be less detrimental to mesenteric blood flow than potent vasoconstrictors. In the absence of active bleeding, systemic anticoagulation is usually indicated, although ideal timing is uncertain and must be individualized.
Definitive management depends on the AMI subtype and on whether peritoneal signs are present. Peritonitis mandates immediate laparotomy with resection of nonviable bowel and source control—no delay for additional testing. For major SMA embolus without peritoneal signs, embolectomy is considered standard, and depending on anatomy and severity, options may include surgical revascularization, catheter-directed thrombolysis/vasodilators, or systemic anticoagulation. For SMA thrombosis, emergent surgical revascularization is traditionally the treatment of choice, though endovascular stenting may be viable in selected patients and is increasingly used. NOMI is treated by reversing the precipitating low-flow/vasoconstrictive state: optimize volume, cardiac output, and oxygen delivery; reduce or stop vasoconstrictors when possible; and consider catheter-directed papaverine (or other vasodilatory/antispasmodic agents) via angiographic techniques. Newer modalities such as two-dimensional perfusion angiography may help assess perfusion response after intraarterial vasodilator therapy. For mesenteric venous thrombosis, management hinges on exam: if peritoneal signs exist, laparotomy and resection are required; if not, immediate heparin anticoagulation followed by longer-term anticoagulation (often warfarin or other long-term strategy) may be sufficient. A major practical limitation of purely percutaneous approaches is that many patients already have nonviable bowel, necessitating laparotomy even if flow is restored.
A planned “second-look” operation is indicated in most patients, typically 24–48 hours after revascularization, to reassess bowel viability and avoid missed progressive ischemia. Chronic/postdischarge care includes close outpatient follow-up. After endovascular treatment, antiplatelet therapy such as clopidogrel for 3–6 months is commonly used along with surveillance for restenosis using duplex ultrasound or CTA. For venous thrombosis, ongoing anticoagulation is required to prevent recurrence, though the optimal duration depends on whether provoking factors or persistent thrombophilia exist.
Prognosis varies by etiology and timing. Outcomes are generally best in mesenteric venous thrombosis and in acute arterial embolism treated promptly, while they remain poor in arterial thrombosis and NOMI, especially when diagnosis is delayed. Delayed recognition leads to infarction with perforation, sepsis, shock, and death. Early surgical consultation is essential, and there should be no delay when peritoneal signs are present; surgery may also be needed diagnostically when clinical suspicion is high and imaging is not immediately available. Prevention focuses on controlling underlying risk factors—especially atherosclerosis prevention (smoking cessation, blood pressure control, statin therapy when indicated) and appropriate management of arrhythmias and hypercoagulable states to reduce recurrence risk.
Don’t-miss points (quick memory anchors): severe pain with minimal early tenderness, high mortality, CTA early, lactate can be late (but >2 suggests necrosis), peritonitis = dead bowel until proven otherwise, early anticoagulation if no bleeding, antibiotics + NG tube, and “second-look” surgery after revascularization.
- Published on
KembaraXtra-Medicine – Acute Lymphoblastic Leukemia (ALL)
Acute lymphoblastic leukemia (ALL) is a malignant disorder of immature lymphoid precursor cells (lymphoblasts) derived from either B-cell or T-cell lineages. It is characterized by uncontrolled proliferation of these malignant lymphoblasts with progressive replacement of normal bone marrow elements, leading to bone marrow failure and peripheral cytopenias. A related entity, lymphoblastic lymphoma (LBL), is diagnosed when disease is primarily extramedullary—most commonly presenting as a mediastinal mass in T-cell disease—and bone marrow involvement is less than 20%. ALL is also called acute lymphocytic leukemia or acute lymphoblastic leukemia and is coded under ICD-10-CM as C91.00 (not having achieved remission), C91.01 (in remission), and C91.02 (in relapse).
ALL is predominantly a disease of children, adolescents, and young adults. The overall incidence is approximately 1.8 per 100,000 persons per year, and about 65% of cases occur in individuals younger than 34 years. It is most commonly diagnosed before age 20 based on SEER data (2015–2019). Incidence varies by race and ethnicity, being more common in Hispanics and whites than in blacks. There is a slight male predominance with a male-to-female ratio of approximately 55:45.
Clinical presentation is typically driven by marrow infiltration and failure, producing symptoms of anemia, thrombocytopenia, and neutropenia. Patients may develop fatigue, pallor, weakness, and shortness of breath from anemia; petechiae, bruising, and bleeding from thrombocytopenia; and fever that may be due to the leukemia itself or secondary infection. Bone pain is common and reflects leukemic infiltration of bones or expansion of the marrow cavity. Many patients have lymphadenopathy or hepatosplenomegaly. Central nervous system involvement may cause neurologic deficits, altered mental status, or other neurologic symptoms. T-cell LBL is classically associated with a mediastinal mass, which can produce respiratory distress and superior vena cava syndrome and requires special precautions because sedation can worsen airway compromise if tracheal compression is present.
Most cases of ALL occur sporadically without clearly established risk factors. Ionizing radiation exposure is a recognized risk factor. Down syndrome (trisomy 21) is associated with an increased leukemia risk of roughly 3% by age 30, predominantly ALL. ALL may also occur in other hereditary premalignancy syndromes such as ataxia-telangiectasia.
The differential diagnosis depends on the clinical context and the presence of lymphocytosis or circulating blasts. In adults with lymphocytosis, disorders such as chronic lymphocytic leukemia, mantle cell lymphoma, marginal zone lymphoma, and hairy cell leukemia must be considered. In adolescents and young adults, infectious mononucleosis syndromes caused by Epstein–Barr virus or cytomegalovirus can cause reactive lymphocyte abnormalities that may appear blast-like. Disorders that may produce circulating blasts or blast-like cells include acute myeloid leukemia, prolymphocytic leukemia, blastoid mantle cell lymphoma, and Burkitt lymphoma (mature B-cell leukemia/lymphoma). Lymphoblastic lymphoma is also closely related and may overlap clinically. Aplastic anemia is an important “don’t miss” alternative, especially because ALL may rarely present without circulating blasts and instead appear as isolated marrow failure.
Diagnosis requires demonstration of an abnormal lymphoblast population and full biologic risk characterization. Flow cytometry is central to lineage assignment and typically identifies B-lineage blasts through CD19 with cytoplasmic CD22 and/or cytoplasmic CD79a, while immature blasts commonly express CD10, CD34, and terminal deoxynucleotidyltransferase (TdT) with absent surface immunoglobulin. Strong CD20 expression, surface immunoglobulin positivity, and TdT negativity should prompt evaluation for mature B-cell neoplasms such as Burkitt leukemia/lymphoma because this distinction critically changes treatment. T-lineage blasts are typically confirmed by cytoplasmic CD3 and CD7. Aberrant myeloid marker expression such as CD13 or CD33 can occur. Early T-cell precursor ALL (ETP-ALL) has a distinct immunologic signature and is clinically important because it carries different prognostic implications and may require different treatment approaches. Cytochemical stains are less specific but may be available quickly; ALL blasts should be negative for myeloperoxidase and esterase stains. A bone marrow examination confirms diagnosis and extent of marrow replacement and provides material for cytogenetic and molecular testing.
Genetic studies are essential because they define major treatment categories, especially Philadelphia chromosome–positive (Ph+) versus Philadelphia chromosome–negative (Ph−) ALL. This distinction is urgent because Ph+ ALL requires incorporation of ABL1 tyrosine kinase inhibitors. Ph+ ALL is uncommon in children (about 2%–5%) but becomes increasingly common with age and represents about 20%–25% of adult ALL overall, rising to more than 50% of cases in patients older than 60 years. Ph status should be determined rapidly via PCR or FISH, ideally within 24–48 hours of diagnosis. The WHO recognizes ALL/LBL variants with recurrent cytogenetic abnormalities as distinct syndromes, including BCR-ABL1 (t(9;22)), KMT2A (MLL) rearrangements (often t(4;11)), ETV6-RUNX1 (t(12;21)), hyperdiploidy, hypodiploidy, IL3-IGH (t(5;14)), TCF3-PBX1 (t(1;19)), iAMP21, and others, along with T-lymphoblastic leukemia/lymphoma and rarer NK lymphoblastic neoplasms. Common abnormalities have prognostic and therapeutic implications: BCR-ABL1 requires a tyrosine kinase inhibitor; KMT2A rearrangements often present with very high white blood cell counts and confer poor prognosis with frequent consideration of transplant; ETV6-RUNX1 and hyperdiploidy generally confer favorable prognosis in children; hypodiploidy confers adverse prognosis; and TCF3-PBX1 has mixed risk implications, including higher CNS relapse risk historically and benefit from intensified antimetabolite therapy in some settings.
Genetic profiling for “Philadelphia-like” (Ph-like) ALL and IKZF1 (IKAROS) alterations can further refine prognosis and may reveal opportunities for targeted therapy. Ph-like ALL resembles Ph+ ALL in expression patterns but lacks BCR-ABL1; it often has kinase or cytokine receptor pathway activation through ABL-class fusions (ABL1, ABL2, CSF1R, PDGFRB) that phenocopy BCR-ABL1 or through CRLF2, JAK2, and EPOR alterations activating JAK/STAT signaling. Other implicated pathways include additional kinases and RAS pathway alterations. These results may affect therapy selection, but testing may not be universally available.
Lumbar puncture is typically performed at diagnosis when feasible to assess for CNS involvement and to initiate CNS prophylaxis. This is particularly important because CNS therapy is required for essentially all patients regardless of whether CNS disease is detected. Before lumbar puncture, coagulation testing is important, especially if thrombocytopenia or disseminated intravascular coagulation is suspected. Imaging evaluation commonly includes a chest radiograph to assess for mediastinal mass and to evaluate fever. CT imaging may be needed for specific symptoms, but contrast exposure should be used cautiously in the setting of suspected tumor lysis–related renal risk.
Initial laboratory evaluation generally demonstrates normocytic anemia and thrombocytopenia; leukocyte counts may be low, normal, or high. Peripheral smear often reveals lymphoblasts, but some patients have marrow-only disease without circulating blasts. Baseline blood work must include assessment of renal and hepatic function (e.g., creatinine and bilirubin), blood glucose because glucocorticoids are core components of therapy, and evaluation for spontaneous tumor lysis syndrome with potassium, calcium, phosphate, and uric acid levels. Tumor lysis syndrome is common in ALL and can be present even before therapy is initiated, making it a major early cause of morbidity and potential mortality. Tumor lysis results from release of intracellular potassium and phosphate and breakdown of nucleic acids into uric acid, which can precipitate renal failure. Hyperkalemia can trigger fatal arrhythmias; hyperphosphatemia promotes calcium phosphate deposition in renal tissue and lowers serum calcium, creating risk of neuromuscular irritability, seizures, and dysrhythmia. Laboratory tumor lysis syndrome is defined by the presence of at least two characteristic metabolic abnormalities within 3 days before or 7 days after therapy, while clinical tumor lysis includes complications such as acute kidney injury, arrhythmia, seizure, or symptomatic hypocalcemia. Corrected calcium is calculated as measured calcium plus 0.8 multiplied by (4 minus measured albumin in g/dL).
Management of tumor lysis syndrome centers on preserving renal function and preventing life-threatening electrolyte complications. Recommended measures include vigorous hydration, often approximating 3 liters of normal saline per day when feasible, with alkalinization not recommended. If urine output is inadequate, forced diuresis may be used to maintain a target urine output near 2 mL/kg/hour, and dialysis is used when potassium, phosphate, volume overload, or renal failure cannot be controlled medically. Allopurinol is routinely used for uric acid prevention, with adult dosing up to 800 mg/day and pediatric dosing typically 300–450 mg/m²/day. Rasburicase rapidly reduces uric acid and is often effective with a single dose at 0.2 mg/kg, but it must be avoided in patients with G6PD deficiency. Phosphate binders are commonly administered though their benefit is uncertain. Asymptomatic hypocalcemia is not treated in order to avoid worsening calcium phosphate deposition.
Modern outcomes for ALL are excellent in children, with survival improving dramatically from about 10% to approximately 90% over the past four decades, representing a major success of contemporary oncology. Adult outcomes remain poorer but have improved substantially, with cure rates around 60% to 70% in standard-risk adult patients in recent trials. Adults have particularly benefited from tyrosine kinase inhibitor therapy for Ph+ ALL, which is more prevalent with age and may represent a large fraction of disease in older adults. Hyperleukocytosis (WBC >100,000/µL) is less common in ALL than in some other leukemias, and high lymphocyte counts can sometimes be tolerated. Prednisone and vincristine often provide rapid cytoreduction, and leukapheresis is rarely required, though it may be used in selected cases.
Treatment of Ph− ALL generally proceeds through four core components. Induction therapy aims to achieve remission and commonly includes corticosteroids, vincristine, an anthracycline such as daunorubicin or doxorubicin, asparaginase, and sometimes cyclophosphamide depending on protocol. In patients whose blasts express CD20 in significant proportion, rituximab has demonstrated benefit and is incorporated into therapy. Consolidation therapy consists of intensive chemotherapy aimed at preventing relapse after remission, commonly using high-dose antimetabolites such as methotrexate and cytarabine in combination with other agents. Maintenance therapy is prolonged low-intensity outpatient therapy usually continued for two to three years after consolidation, frequently using the POMP backbone of prednisone, monthly vincristine, methotrexate, and oral 6-mercaptopurine. CNS prophylaxis is universal and typically delivered via intrathecal therapy—methotrexate alone or combined with cytarabine and hydrocortisone—administered through lumbar puncture or an Ommaya reservoir. Cranial radiotherapy is generally avoided due to toxicity and is reserved for high-risk situations such as active CNS disease at diagnosis.
Protocol selection varies by institution and trial availability. In the modern era, adolescents and young adults (often defined as ages 15–39) are frequently treated using pediatric or pediatric-inspired regimens because outcomes historically were superior compared with traditional adult regimens. This has influenced practice patterns such that many younger AYA patients are treated on pediatric-style protocols, either within pediatric services or with adult programs using pediatric-inspired approaches.
Allogeneic hematopoietic stem cell transplantation (HSCT) in first remission remains controversial because chemotherapy outcomes have improved. In general, HSCT is considered when predicted cure likelihood with chemotherapy alone is below roughly 50% to 60%, factoring in age, genetics, minimal residual disease status, and donor availability. Autologous transplantation is rarely used in Ph− ALL and has not shown benefit as a replacement strategy. Large comparative data have shown that allogeneic transplant in first complete remission reduces relapse risk compared with chemotherapy or autologous transplant, and can improve overall survival in standard-risk patients, but treatment-related mortality is higher in older and high-risk groups and can offset relapse reduction benefits.
Risk assessment is increasingly driven by biologic markers and response assessment rather than older clinical groupings alone. High-risk features in contemporary trials include KMT2A (MLL) rearrangements, hypodiploidy, persistent minimal residual disease after induction or consolidation, Ph-like genomic signature in Ph− disease, and early T-cell precursor ALL immunophenotype features such as absent CD1a and CD8, weak CD5, and presence of myeloid or stem-cell antigen expression. Prognosis is also influenced by age, presenting white blood cell count, CNS or testicular involvement, cytogenetic abnormalities, and MRD dynamics. MRD is among the strongest predictors: end-of-induction MRD below 0.01% is associated with excellent outcomes, whereas persistent MRD after consolidation predicts very poor prognosis and often leads to consideration of transplant in first remission.
Therapy for Ph+ ALL incorporates an ABL tyrosine kinase inhibitor such as imatinib, dasatinib, nilotinib, or ponatinib combined with chemotherapy. Reported two-year survival rates in Ph+ disease are approximately 50% to 65% across regimens. Some approaches use reduced-intensity induction such as dasatinib with prednisone or imatinib with vincristine and prednisone, achieving very high remission rates while reducing early toxicity and hospitalization. Allogeneic transplantation is often used as consolidation when feasible, though this strategy is increasingly debated with modern TKI-based regimens. Maintenance TKI therapy is typically continued after transplant or after non-transplant therapy.
Relapsed ALL remains challenging. Allogeneic transplantation is often offered, but relapse after transplant is common and long-term cure rates have historically been low near 20%. Newer immunotherapies have transformed outcomes, especially in B-cell ALL. CD19-directed CAR-T therapy has produced very high remission rates in pediatric and young adult patients, with substantial proportions maintaining durable remission at six months, though severe toxicity can occur. Cytokine release syndrome is a major risk and can manifest with vascular leak, hypotension, respiratory and renal failure, coagulopathy, and is treated with tocilizumab, an IL-6 receptor–blocking antibody. Tisagenlecleucel (Kymriah) is a CAR-T product used in pediatric and young adult B-cell ALL, while brexucabtagene autoleucel (Tecartus) is approved for adult relapsed or refractory B-cell ALL based on trials showing meaningful complete response rates, though cytokine release syndrome and neurotoxicity are frequent. Blinatumomab is a bispecific T-cell engager binding CD19 and CD3 that redirects T cells toward leukemia cells and is given as continuous infusion in cycles; it improves remission rates compared with chemotherapy in relapsed settings but responses are not always durable and cytokine release syndrome can occur. Inotuzumab ozogamicin is an anti-CD22 antibody-drug conjugate that produces high remission rates and can bridge patients to transplant, though median remission durations may be limited.
Survivorship care is increasingly important because tens of thousands of childhood and adult ALL survivors now live long-term and are regularly encountered in general practice. Late effects include secondary malignancies related to chemotherapy (often within 5–10 years) and radiation (risk may persist without plateau), cardiomyopathy and heart failure from anthracycline exposure that may emerge decades later, osteopenia and avascular necrosis from glucocorticoid therapy, obesity and metabolic derangements, and neurocognitive deficits. Long-term follow-up commonly includes periodic echocardiography every 3–5 years for asymptomatic cardiomyopathy surveillance, performed more frequently in those with higher anthracycline exposure. Survivors also require screening for malignancy and endocrine disorders in relevant radiation fields if radiation was used, along with proactive monitoring and counseling for obesity and cardiometabolic risk.

- Published on
KembaraXtra-Medicine – Acute Lower Gastrointestinal Bleeding
Acute lower gastrointestinal (GI) bleeding is traditionally defined as bleeding that originates distal to the ligament of Treitz, though modern definitions more specifically restrict the source to the colon or rectum. It most commonly presents as hematochezia, which may appear as bright red or maroon blood per rectum, and less commonly as melena when bleeding originates from the proximal colon or small bowel. The condition is also referred to as acute colonic bleeding or gastrointestinal hemorrhage and is classified under ICD-10-CM codes K62.5, K92.1, and K92.2.
The annual incidence of hospitalization for acute lower GI bleeding in the United States ranges from approximately 33 to 87 per 100,000 individuals, representing about half the incidence of upper GI bleeding. In-hospital mortality ranges from 2.5% to 3.9%. Despite relatively low short-term mortality, recurrence is common, with rebleeding rates of 13% to 19% at one year and up to 46% at five years. The condition disproportionately affects older adults, with incidence increasing dramatically with age, particularly beyond 65 years. Elderly patients also experience higher in-hospital mortality and worse overall outcomes. Men are affected more frequently than women.
Multiple risk factors contribute to the development of acute lower GI bleeding. These include the use of antithrombotic agents, NSAIDs, aspirin, and alcohol; the presence of gastrointestinal malignancy, atrial fibrillation, coagulopathies, prior GI bleeding, cirrhosis, constipation, congenital malformations, inflammatory bowel disease, and prior radiation exposure. Additional risk factors include recent infectious illness, recent travel, abdominal aortic aneurysm repair, and portal hypertension. Among etiologies, diverticulosis is the most common cause, accounting for approximately 30% of cases, followed by internal hemorrhoids. Other causes include ischemic, infectious, or inflammatory colitis; angioectasia; rectal ulcers; postpolypectomy bleeding; neoplasms; vascular malformations; and anorectal disease. Importantly, up to 15% of patients presenting with apparent lower GI bleeding are ultimately found to have an upper GI source with rapid intestinal transit.
Clinical assessment begins with careful evaluation of hemodynamic stability and a focused history aimed at identifying the bleeding source and underlying cause. Weight loss and abdominal pain raise concern for inflammatory bowel disease or malignancy. A history of recent abdominal aortic aneurysm repair, particularly when preceded by a “sentinel bleed,” should prompt immediate concern for an aortoenteric fistula. Patients with cirrhosis may bleed from portal hypertensive sources such as colonic or rectal varices. Bright red blood per rectum typically suggests distal colonic or anorectal bleeding but may also occur in brisk upper GI hemorrhage, whereas melena generally originates from the upper GI tract, small bowel, or proximal colon. A detailed medication review is essential, particularly for NSAIDs, anticoagulants, antiplatelet agents, and beta blockers, which may blunt compensatory tachycardia.
Physical examination should include assessment of vital signs, including orthostatic measurements if the patient is stable. Clinicians should evaluate for pallor, delayed capillary refill, and signs of hypovolemia, as well as stigmata of chronic liver disease such as jaundice and telangiectasias. Abdominal examination should assess for tenderness, masses, bowel sounds, and hepatosplenomegaly, with absent bowel sounds raising concern for perforation. A rectal examination is mandatory and should document the presence, color, and volume of blood, as well as masses, fissures, hemorrhoids, tenderness, or skin changes. If no gross blood is visible, fecal occult testing should be performed. It is critical to exclude non-GI sources of bleeding, including hematuria, vaginal bleeding, and external wounds.
Initial management prioritizes airway, breathing, and circulation, with rapid establishment of large-bore intravenous access and prompt volume resuscitation. Crystalloid boluses should be administered to maintain a systolic blood pressure of at least 100 mm Hg. Blood transfusion is recommended for hemoglobin levels of 7 g/dL or less, or 8 g/dL in patients older than 65 years or those with coronary artery disease or multiple comorbidities. Platelet counts should be maintained above 50,000/µL, and the INR should be corrected to 1.5 or less when feasible. Antithrombotic agents and NSAIDs should be discontinued when possible, in consultation with the prescribing physician.
Laboratory evaluation includes serial complete blood counts to monitor hemoglobin trends, comprehensive metabolic panels to assess renal function and liver disease, coagulation studies including INR and partial thromboplastin time, and blood typing and crossmatching in anticipation of transfusion. Stool studies may be indicated when infectious colitis is suspected, and inflammatory markers may assist in identifying inflammatory or ischemic etiologies.
Imaging plays a key role in diagnosis. CT angiography is highly sensitive and specific for identifying active bleeding and abnormal vasculature and is often the preferred initial imaging modality in unstable patients. If CT angiography is nondiagnostic, a tagged red blood cell scan may be performed. CT imaging of the abdomen and pelvis may identify malignancy or other structural pathology. Plain abdominal or chest radiographs are indicated when perforation or foreign body ingestion is suspected. Ultrasound may be useful for suspected intussusception, and a technetium-99 Meckel scan is indicated when Meckel diverticulum is suspected, particularly in younger patients.
Colonoscopy remains the cornerstone of diagnosis and therapy once the patient is stabilized. It allows direct visualization of the colonic mucosa and enables therapeutic interventions such as clipping, cauterization, injection therapy, or band ligation. Anoscopy may be performed when anorectal sources such as hemorrhoids are suspected. Endoscopy is indicated when an upper GI source is suspected. Interventional radiology may be required for embolization in cases of ongoing bleeding, and surgical intervention is reserved for refractory bleeding, aortoenteric fistula, failed nonoperative management, or complications such as perforation. Balloon tamponade may be used in select cases for severe esophageal or anorectal bleeding.
Pharmacologic therapy includes proton pump inhibitors or histamine-2 receptor blockers when an upper GI source with rapid transit is suspected. Chronic management focuses on treating the underlying cause, avoiding NSAIDs and alcohol, correcting coagulopathies, and managing portal hypertension when present. Many patients are discharged on proton pump inhibitors. Fiber supplementation, stool softeners, and topical analgesic agents may reduce symptoms and recurrence of hemorrhoidal bleeding.
Most cases of acute lower GI bleeding resolve spontaneously. Hemodynamically stable patients without significant anemia or ongoing bleeding may be managed as outpatients with close follow-up by primary care or gastroenterology. Patients with hemodynamic compromise, ongoing hemorrhage, shock, or significant comorbidities require hospital admission, often to an intensive care unit, for close monitoring and urgent intervention. Clinical predictors of severe bleeding include aspirin use, the presence of two or more comorbidities, heart rate greater than 100 beats per minute, and systolic blood pressure less than 115 mm Hg. Overall mortality ranges from 2.4% to 3.9%, with age greater than 65 years, intestinal ischemia, and multiple comorbidities serving as independent predictors of in-hospital mortality. All patients should be counseled regarding the high risk of rebleeding and the importance of prompt evaluation if symptoms recur, with referral to gastroenterology for follow-up care.
- Published on
KembaraXtra-Medicine- Acute Liver Failure (ALF)
Acute liver failure (ALF) is the rapid onset of severe liver dysfunction causing coagulopathy and altered mentation in a patient without previously known liver disease. In practice, it is defined by acute severe hepatic injury lasting <26 weeks, synthetic dysfunction with inr>1.5, and any degree of hepatic encephalopathy in the absence of preexisting cirrhosis and excluding acute alcoholic hepatitis. ALF can also be diagnosed in certain patients with preexisting liver disorders such as Wilson disease, vertically acquired hepatitis B, or autoimmune hepatitis, as long as the diagnosis was made within the preceding 26 weeks. ALF must be distinguished from acutely decompensated cirrhosis (new ascites, encephalopathy, and/or GI bleeding in established cirrhosis) and acute-on-chronic liver failure (acute deterioration in chronic liver disease with intense systemic inflammation and potential multiorgan failure).
Several classification systems describe ALF based on timing of encephalopathy relative to symptom onset or jaundice. The British scheme categorizes by interval between jaundice and encephalopathy (hyperacute, acute, subacute, late-onset), and other systems (French classification, International Association for the Study of Acute Liver Failure) similarly emphasize time course, recognizing that more rapid presentations may behave differently clinically. Common synonyms include fulminant hepatic failure, fulminant hepatitis, fulminant hepatic necrosis, and acute hepatic necrosis. ICD-10-CM codes include K72, K72.0, K72.00 (without coma), and K72.01 (with coma).
ALF is uncommon but high-stakes. Reported incidence is about 2000 cases per year in the U.S. and 1 to 8 per million population in the U.K. It is described as occurring more often in women and often affecting younger individuals. Risk factors include intentional or unintentional drug overdose, exposures increasing risk of viral hepatitis (IV drug use, blood/body fluid exposure, transfusions, hemodialysis, intranasal cocaine use, imprisonment, travel to endemic regions), prior alcohol use, hepatotoxic medications, and critical illness.
Symptoms are often nonspecific early and vary by cause. Patients may develop fatigue, lethargy, anorexia, nausea/vomiting, pruritus, jaundice, and right upper quadrant pain or distention. More severe presentations can include hypotension, sepsis, and encephalopathy. Physical examination, by definition, includes neurologic abnormalities of encephalopathy, and may show jaundice, asterixis, hepatomegaly or decreased liver span, and ascites. In advanced cases, cerebral edema and increased intracranial pressure can occur, with concerning findings such as pupillary abnormalities, hypertension with bradycardia and respiratory depression (Cushing triad), loss of brainstem reflexes, and seizures. Vesicular skin lesions raise concern for HSV, and a family history of unexplained liver disease should prompt slit-lamp exam for Kayser–Fleischer rings suggestive of Wilson disease.
In the Western world, the most common causes include acetaminophen toxicity (~46%), indeterminate causes, idiosyncratic drug reactions, and viral hepatitis (especially hepatitis A and B). Other causes include autoimmune hepatitis, Wilson disease, ischemic hepatopathy (“shock liver”), Budd–Chiari syndrome, acute fatty liver of pregnancy, veno-occlusive disease, toxic ingestions such as Amanita phalloides mushroom poisoning, sepsis, malignant hepatic infiltration, and viral etiologies beyond hepatitis viruses (e.g., adenovirus, hepatitis E, HSV).
Laboratory findings reflect loss of synthetic and metabolic function. Coagulopathy is typical due to reduced hepatic synthesis of clotting factors (II, V, VII, IX, X), producing INR >1.5. Transaminases and bilirubin are usually elevated, and thrombocytopenia may occur. Many patients develop acute kidney injury (often cited around 30%–50%), and hypoglycemia can occur from impaired gluconeogenesis. Electrolyte disturbances are common (hyponatremia, hypophosphatemia, hypomagnesemia, hypokalemia), along with metabolic acidosis or respiratory alkalosis, elevated lactate dehydrogenase, and elevated ammonia.
The differential diagnosis includes severe acute hepatitis/acute liver injury (jaundice and coagulopathy without encephalopathy), acute-on-chronic liver failure, decompensated cirrhosis, and hepatocellular carcinoma. Distinguishing ALF from portal-systemic encephalopathy due to chronic liver disease can be aided by the clinical context: ALF is typically acute, with very high transaminases, a small tender liver, and absence of chronic stigmata early, whereas chronic disease often has cachexia, collateral circulation, ascites, and less dramatic transaminase elevation.
Workup relies on rapid severity assessment and urgent search for etiology, often with limited history due to encephalopathy. Clinicians should obtain collateral history regarding medications (including OTC and herbal agents), alcohol and recreational drugs, onset of jaundice and symptoms, psychiatric history (including self-harm), travel and exposure risks for viral hepatitis, transfusions, family history of liver disease, malignancy history, and hypercoagulable conditions. Examination should assess mental status and grade encephalopathy (from mild behavioral/sleep changes through coma). Laboratory evaluation typically includes CBC, complete liver panel and INR/PT, comprehensive metabolic panel with glucose and electrolytes, arterial blood gas and lactate, blood type and screen, acetaminophen level, ethanol level and toxicology screen, hepatitis serologies and viral loads where appropriate, HSV testing (often PCR), EBV/CMV testing, ceruloplasmin (with copper studies if suspicion is high), pregnancy test, ammonia, autoimmune markers (ANA, anti–smooth muscle, anti-LKM1, total IgG), and HIV testing. Imaging usually includes abdominal ultrasound with Doppler to assess for Budd–Chiari, congestion, portal hypertension, and steatosis, with consideration of cross-sectional liver imaging when malignancy or infiltrative disease is possible. Head CT or MRI may be used to evaluate alternative causes of altered mental status. A transjugular liver biopsy is considered when the cause remains unclear or when autoimmune hepatitis, malignancy, or HSV is strongly suspected.
Complications include cerebral edema (reported up to ~40% in some series), hypoglycemia, lactic acidosis, acute kidney injury, pancreatitis (notably with acetaminophen), infections due to impaired immune function, high-output cardiac failure, ARDS, and hypotension from poor intake and third spacing.
Treatment focuses on rapid stabilization, supportive ICU-level care when indicated, targeted therapy for the underlying cause, and early transplant evaluation. Encephalopathy grade guides level of care: grade I may be managed on a medical ward with frequent neuro checks, while grades II–IV generally require ICU management, head-of-bed elevation, and for grades III–IV, intubation and mechanical ventilation. Sedatives should be minimized because they can accumulate and obscure neurologic assessment; a low-stimulation environment is recommended. Early notification of a liver specialist and early transfer to a transplant center are essential because deterioration can later make transport unsafe.
Supportive care includes early nutrition (to prevent catabolism) and careful fluid management with crystalloids tailored to hemodynamics and metabolic needs, including dextrose-containing fluids if hypoglycemia is present. If intracranial hypertension develops, mannitol can transiently reduce cerebral edema; sodium is often maintained in a higher range (e.g., 140–150 mmol/L), and hypertonic saline may be used in highest-risk patients. Prophylactic hyperventilation is not recommended. Seizures may be treated with agents such as phenytoin; short-acting benzodiazepines can be used if necessary, but sedation is generally avoided when possible. Lactulose and rifaximin are not routinely recommended for encephalopathy in ALF, and neomycin should be avoided due to nephrotoxicity risk.
Infection management is critical: broad-spectrum antibiotics are started when infection is suspected, but routine empiric antibiotics without suspicion are not supported; antifungals may be added if there is poor response or high concern. Hemodynamic support may require norepinephrine as first-line vasopressor, with vasopressin as an adjunct; refractory hypotension should prompt evaluation for adrenal insufficiency. If renal replacement therapy is required, continuous modalities are often preferred over intermittent hemodialysis in unstable patients. Stress ulcer prophylaxis is recommended. Routine correction of INR or thrombocytopenia is not recommended without bleeding, but vitamin K is generally given because deficiency is common; plasma or clotting factors are reserved for active bleeding or high-risk procedures.
Cause-specific therapy is crucial, especially for acetaminophen toxicity. There should be a low threshold to start IV N-acetylcysteine (NAC) when acetaminophen overdose is known or suspected because it is beneficial and generally safe. A commonly used 20-hour IV regimen is a loading dose followed by staged infusions, with reassessment of acetaminophen level and ALT near the end of the protocol and extension of therapy if levels remain elevated or enzymes are rising. NAC use in non–acetaminophen ALF remains controversial and is center-specific, with inconclusive evidence for routine benefit.
Bridging and advanced therapies include extracorporeal liver support systems and high-volume plasma exchange in select contexts, but these are not routinely recommended in guidelines as definitive therapy; they may be used as a bridge to recovery or transplant in specialized centers. For patients with high-grade encephalopathy awaiting transplant, some centers use intracranial pressure monitoring when expertise is available; otherwise, frequent neurologic exams are essential. Coagulation tests and metabolic panels are monitored frequently, while avoiding unnecessary correction of INR because it interferes with prognostication.
Prognosis has improved over time but remains serious, with overall mortality often cited around 30%–40%. Outcomes vary by etiology: transplant-free survival is generally better with acetaminophen-related ALF, hepatitis A, ischemic (“shock”) liver, and pregnancy-related disease, and worse with many other causes. Higher-grade encephalopathy and renal dysfunction predict worse outcomes in non–acetaminophen ALF. Prognostic models include King’s College criteria, Clichy criteria, MELD, and APACHE II; King’s College criteria are widely used to guide transplant referral. Liver transplantation provides definitive therapy for many patients meeting criteria, though contraindications include irreversible brain injury, uncontrolled sepsis, severe comorbidities including malignancy, escalating dependence on ventilator/inotropes, active substance abuse, severe uncontrolled psychiatric illness affecting adherence, and inadequate social support.
Key practical points are that ALF requires INR >1.5 plus encephalopathy within 26 weeks in a patient without known chronic liver disease, acetaminophen accounts for a large portion of cases, early transfer to a transplant center can be lifesaving, NAC should be started early when acetaminophen is possible, and coagulopathy should not be corrected unless bleeding is present or a high-risk procedure is necessary.
Acute liver failure (ALF) is the rapid onset of severe liver dysfunction causing coagulopathy and altered mentation in a patient without previously known liver disease. In practice, it is defined by acute severe hepatic injury lasting <26 weeks, synthetic dysfunction with inr>1.5, and any degree of hepatic encephalopathy in the absence of preexisting cirrhosis and excluding acute alcoholic hepatitis. ALF can also be diagnosed in certain patients with preexisting liver disorders such as Wilson disease, vertically acquired hepatitis B, or autoimmune hepatitis, as long as the diagnosis was made within the preceding 26 weeks. ALF must be distinguished from acutely decompensated cirrhosis (new ascites, encephalopathy, and/or GI bleeding in established cirrhosis) and acute-on-chronic liver failure (acute deterioration in chronic liver disease with intense systemic inflammation and potential multiorgan failure).
Several classification systems describe ALF based on timing of encephalopathy relative to symptom onset or jaundice. The British scheme categorizes by interval between jaundice and encephalopathy (hyperacute, acute, subacute, late-onset), and other systems (French classification, International Association for the Study of Acute Liver Failure) similarly emphasize time course, recognizing that more rapid presentations may behave differently clinically. Common synonyms include fulminant hepatic failure, fulminant hepatitis, fulminant hepatic necrosis, and acute hepatic necrosis. ICD-10-CM codes include K72, K72.0, K72.00 (without coma), and K72.01 (with coma).
ALF is uncommon but high-stakes. Reported incidence is about 2000 cases per year in the U.S. and 1 to 8 per million population in the U.K. It is described as occurring more often in women and often affecting younger individuals. Risk factors include intentional or unintentional drug overdose, exposures increasing risk of viral hepatitis (IV drug use, blood/body fluid exposure, transfusions, hemodialysis, intranasal cocaine use, imprisonment, travel to endemic regions), prior alcohol use, hepatotoxic medications, and critical illness.
Symptoms are often nonspecific early and vary by cause. Patients may develop fatigue, lethargy, anorexia, nausea/vomiting, pruritus, jaundice, and right upper quadrant pain or distention. More severe presentations can include hypotension, sepsis, and encephalopathy. Physical examination, by definition, includes neurologic abnormalities of encephalopathy, and may show jaundice, asterixis, hepatomegaly or decreased liver span, and ascites. In advanced cases, cerebral edema and increased intracranial pressure can occur, with concerning findings such as pupillary abnormalities, hypertension with bradycardia and respiratory depression (Cushing triad), loss of brainstem reflexes, and seizures. Vesicular skin lesions raise concern for HSV, and a family history of unexplained liver disease should prompt slit-lamp exam for Kayser–Fleischer rings suggestive of Wilson disease.
In the Western world, the most common causes include acetaminophen toxicity (~46%), indeterminate causes, idiosyncratic drug reactions, and viral hepatitis (especially hepatitis A and B). Other causes include autoimmune hepatitis, Wilson disease, ischemic hepatopathy (“shock liver”), Budd–Chiari syndrome, acute fatty liver of pregnancy, veno-occlusive disease, toxic ingestions such as Amanita phalloides mushroom poisoning, sepsis, malignant hepatic infiltration, and viral etiologies beyond hepatitis viruses (e.g., adenovirus, hepatitis E, HSV).
Laboratory findings reflect loss of synthetic and metabolic function. Coagulopathy is typical due to reduced hepatic synthesis of clotting factors (II, V, VII, IX, X), producing INR >1.5. Transaminases and bilirubin are usually elevated, and thrombocytopenia may occur. Many patients develop acute kidney injury (often cited around 30%–50%), and hypoglycemia can occur from impaired gluconeogenesis. Electrolyte disturbances are common (hyponatremia, hypophosphatemia, hypomagnesemia, hypokalemia), along with metabolic acidosis or respiratory alkalosis, elevated lactate dehydrogenase, and elevated ammonia.
The differential diagnosis includes severe acute hepatitis/acute liver injury (jaundice and coagulopathy without encephalopathy), acute-on-chronic liver failure, decompensated cirrhosis, and hepatocellular carcinoma. Distinguishing ALF from portal-systemic encephalopathy due to chronic liver disease can be aided by the clinical context: ALF is typically acute, with very high transaminases, a small tender liver, and absence of chronic stigmata early, whereas chronic disease often has cachexia, collateral circulation, ascites, and less dramatic transaminase elevation.
Workup relies on rapid severity assessment and urgent search for etiology, often with limited history due to encephalopathy. Clinicians should obtain collateral history regarding medications (including OTC and herbal agents), alcohol and recreational drugs, onset of jaundice and symptoms, psychiatric history (including self-harm), travel and exposure risks for viral hepatitis, transfusions, family history of liver disease, malignancy history, and hypercoagulable conditions. Examination should assess mental status and grade encephalopathy (from mild behavioral/sleep changes through coma). Laboratory evaluation typically includes CBC, complete liver panel and INR/PT, comprehensive metabolic panel with glucose and electrolytes, arterial blood gas and lactate, blood type and screen, acetaminophen level, ethanol level and toxicology screen, hepatitis serologies and viral loads where appropriate, HSV testing (often PCR), EBV/CMV testing, ceruloplasmin (with copper studies if suspicion is high), pregnancy test, ammonia, autoimmune markers (ANA, anti–smooth muscle, anti-LKM1, total IgG), and HIV testing. Imaging usually includes abdominal ultrasound with Doppler to assess for Budd–Chiari, congestion, portal hypertension, and steatosis, with consideration of cross-sectional liver imaging when malignancy or infiltrative disease is possible. Head CT or MRI may be used to evaluate alternative causes of altered mental status. A transjugular liver biopsy is considered when the cause remains unclear or when autoimmune hepatitis, malignancy, or HSV is strongly suspected.
Complications include cerebral edema (reported up to ~40% in some series), hypoglycemia, lactic acidosis, acute kidney injury, pancreatitis (notably with acetaminophen), infections due to impaired immune function, high-output cardiac failure, ARDS, and hypotension from poor intake and third spacing.
Treatment focuses on rapid stabilization, supportive ICU-level care when indicated, targeted therapy for the underlying cause, and early transplant evaluation. Encephalopathy grade guides level of care: grade I may be managed on a medical ward with frequent neuro checks, while grades II–IV generally require ICU management, head-of-bed elevation, and for grades III–IV, intubation and mechanical ventilation. Sedatives should be minimized because they can accumulate and obscure neurologic assessment; a low-stimulation environment is recommended. Early notification of a liver specialist and early transfer to a transplant center are essential because deterioration can later make transport unsafe.
Supportive care includes early nutrition (to prevent catabolism) and careful fluid management with crystalloids tailored to hemodynamics and metabolic needs, including dextrose-containing fluids if hypoglycemia is present. If intracranial hypertension develops, mannitol can transiently reduce cerebral edema; sodium is often maintained in a higher range (e.g., 140–150 mmol/L), and hypertonic saline may be used in highest-risk patients. Prophylactic hyperventilation is not recommended. Seizures may be treated with agents such as phenytoin; short-acting benzodiazepines can be used if necessary, but sedation is generally avoided when possible. Lactulose and rifaximin are not routinely recommended for encephalopathy in ALF, and neomycin should be avoided due to nephrotoxicity risk.
Infection management is critical: broad-spectrum antibiotics are started when infection is suspected, but routine empiric antibiotics without suspicion are not supported; antifungals may be added if there is poor response or high concern. Hemodynamic support may require norepinephrine as first-line vasopressor, with vasopressin as an adjunct; refractory hypotension should prompt evaluation for adrenal insufficiency. If renal replacement therapy is required, continuous modalities are often preferred over intermittent hemodialysis in unstable patients. Stress ulcer prophylaxis is recommended. Routine correction of INR or thrombocytopenia is not recommended without bleeding, but vitamin K is generally given because deficiency is common; plasma or clotting factors are reserved for active bleeding or high-risk procedures.
Cause-specific therapy is crucial, especially for acetaminophen toxicity. There should be a low threshold to start IV N-acetylcysteine (NAC) when acetaminophen overdose is known or suspected because it is beneficial and generally safe. A commonly used 20-hour IV regimen is a loading dose followed by staged infusions, with reassessment of acetaminophen level and ALT near the end of the protocol and extension of therapy if levels remain elevated or enzymes are rising. NAC use in non–acetaminophen ALF remains controversial and is center-specific, with inconclusive evidence for routine benefit.
Bridging and advanced therapies include extracorporeal liver support systems and high-volume plasma exchange in select contexts, but these are not routinely recommended in guidelines as definitive therapy; they may be used as a bridge to recovery or transplant in specialized centers. For patients with high-grade encephalopathy awaiting transplant, some centers use intracranial pressure monitoring when expertise is available; otherwise, frequent neurologic exams are essential. Coagulation tests and metabolic panels are monitored frequently, while avoiding unnecessary correction of INR because it interferes with prognostication.
Prognosis has improved over time but remains serious, with overall mortality often cited around 30%–40%. Outcomes vary by etiology: transplant-free survival is generally better with acetaminophen-related ALF, hepatitis A, ischemic (“shock”) liver, and pregnancy-related disease, and worse with many other causes. Higher-grade encephalopathy and renal dysfunction predict worse outcomes in non–acetaminophen ALF. Prognostic models include King’s College criteria, Clichy criteria, MELD, and APACHE II; King’s College criteria are widely used to guide transplant referral. Liver transplantation provides definitive therapy for many patients meeting criteria, though contraindications include irreversible brain injury, uncontrolled sepsis, severe comorbidities including malignancy, escalating dependence on ventilator/inotropes, active substance abuse, severe uncontrolled psychiatric illness affecting adherence, and inadequate social support.
Key practical points are that ALF requires INR >1.5 plus encephalopathy within 26 weeks in a patient without known chronic liver disease, acetaminophen accounts for a large portion of cases, early transfer to a transplant center can be lifesaving, NAC should be started early when acetaminophen is possible, and coagulopathy should not be corrected unless bleeding is present or a high-risk procedure is necessary.

- Published on
KembaraXtra-Medicine- Acute Disseminated Encephalomyelitis (ADEM)
Acute disseminated encephalomyelitis (ADEM) is an acquired inflammatory demyelinating syndrome of the central nervous system (CNS), most often occurring in children and frequently considered postinfectious in origin. It presents with acute-onset, widespread CNS inflammation and demyelination. Current diagnostic criteria emphasize the combination of acute polyfocal neurologic deficits with encephalopathy (a required core feature) and supportive brain MRI findings showing multifocal areas of increased T2/FLAIR signal.
ADEM is typically a monophasic illness, and more than 90% of children experience only a single event. Some children may develop evolving or new neurologic deficits within 90 days of symptom onset; this is still considered part of the same ADEM event rather than a relapse. When new attacks occur more than 90 days after onset, they may represent multiphasic ADEM (MDEM) or another demyelinating syndrome such as optic neuritis or transverse myelitis. A more fulminant variant, acute hemorrhagic leukoencephalitis (AHLE), can resemble ADEM clinically but progresses more rapidly and is associated with multifocal hemorrhages on imaging.
Epidemiologically, ADEM is most prevalent in school-aged children, with a mean age of onset around 5 to 8 years, and a slight male predominance. Incidence is estimated at 0.1 to 0.6 per 100,000 per year, with peak occurrence in winter and spring. Because ADEM is generally transient, prevalence estimates are not well established. Although ADEM can occur in adults, adult-onset cases tend to have a poorer prognosis than pediatric cases.
Clinically, many patients have a febrile illness in the weeks before neurologic symptoms, often an upper respiratory infection. Children may present with headache, vomiting, vision changes, gait disturbance, meningismus, and seizures (reported in about one-third of patients). The hallmark feature is encephalopathy, ranging from irritability and confusion to lethargy and coma. The spinal cord may be involved (transverse myelitis), producing motor and sensory deficits and possible bowel or bladder dysfunction, and cranial neuropathies—including optic neuritis—may occur. The course can be rapidly progressive, and severe cases may require intensive care monitoring.
The etiology is strongly linked temporally to recent infections, and molecular mimicry has been proposed as a mechanism, though definitive proof is lacking. ADEM has been reported following various infections, and case reports have described ADEM after COVID-19 infection; however, a causal role for SARS-CoV-2 has not been established. Importantly, recent comprehensive studies have not shown associations between ADEM and common vaccines, and while rare reports exist after SARS-CoV-2 mRNA vaccination, no causal relationship has been demonstrated.
Diagnosis is clinical and requires encephalopathy plus polyfocal deficits; MRI abnormalities alone are not sufficient in the absence of the key clinical features. The differential diagnosis includes neuromyelitis optica spectrum disorder (NMOSD) associated with aquaporin-4 antibodies, MOG-associated disease (MOGAD), multiple sclerosis (MS), meningitis/encephalitis, brain mass lesions, CNS vasculitis, AHLE, hemophagocytic lymphohistiocytosis, and certain metabolic or mitochondrial disorders. Although ADEM can rarely be the first event of MS, MS criteria require evidence of dissemination in time and space (often supported by CSF oligoclonal bands) and typically a non-ADEM clinical pattern.
Workup generally includes evaluation for infectious meningoencephalitis when clinically indicated, cerebrospinal fluid (CSF) analysis, and targeted serum testing. CSF often shows pleocytosis reflecting inflammation. Oligoclonal bands that are present in CSF but not serum are uncommon in ADEM and, while nonspecific, are more typically associated with MS. Serum testing for MOG-IgG is important because approximately half of children with ADEM have MOG antibodies at presentation, and most cases of multiphasic ADEM are MOG-antibody positive. MOG testing should be performed in serum (live cell-based assays with clearly positive titers are preferred), because CSF testing is less specific for MOGAD. Testing for AQP4-IgG is used to identify NMOSD, which is usually a chronic relapsing disease with potentially poor recovery without appropriate long-term management.
Neuroimaging is central to evaluation. Head CT is often performed initially to exclude mass lesions but can miss demyelinating disease due to low sensitivity for white matter changes. Brain and spine MRI with and without contrast is recommended and commonly shows disseminated T2/FLAIR hyperintense lesions across supratentorial and infratentorial white matter, cortex, deep gray matter, brainstem, optic nerves, and sometimes the spinal cord. Lesions are often bilateral, large, poorly demarcated, and only a minority enhance with gadolinium. Follow-up MRI after monophasic ADEM typically demonstrates improvement or resolution over subsequent months.
Treatment is focused on rapid anti-inflammatory therapy and supportive care. Many patients—especially those with severe encephalopathy, seizures, brainstem involvement, or respiratory compromise—require ICU monitoring. First-line therapy is high-dose intravenous corticosteroids, commonly methylprednisolone 30 mg/kg/day (maximum 1000 mg/day) for 3 to 5 days. If there is inadequate clinical improvement, second-line therapies include intravenous immunoglobulin (IVIG) totaling 2 g/kg divided over 2 to 5 days, or 5 to 7 plasma exchanges.
Most patients improve with short-course corticosteroids and do not require a prolonged steroid taper. Long-term steroids are not indicated, even when MOG antibodies are present. Chronic immunomodulatory therapy is reserved for patients who are diagnosed with relapsing conditions such as NMOSD, relapsing MOGAD, or MS, and should be guided by neuroimmunology specialists. A single MOG-related demyelinating event generally does not require chronic immunotherapy, and currently there are no reliable biomarkers that predict which MOGAD patients will relapse.
Disposition depends on severity; significant encephalopathy, seizures, increased intracranial pressure concerns, or ventilatory dysfunction warrant inpatient—and often ICU—care until stabilization. Neurology consultation is recommended for diagnosis, treatment planning, and follow-up. Clinically, the appearance of relapses or new lesions beyond the expected monophasic window is not consistent with monophasic ADEM and should prompt reassessment for multiphasic ADEM, MOGAD, NMOSD, or MS.
Acute disseminated encephalomyelitis (ADEM) is an acquired inflammatory demyelinating syndrome of the central nervous system (CNS), most often occurring in children and frequently considered postinfectious in origin. It presents with acute-onset, widespread CNS inflammation and demyelination. Current diagnostic criteria emphasize the combination of acute polyfocal neurologic deficits with encephalopathy (a required core feature) and supportive brain MRI findings showing multifocal areas of increased T2/FLAIR signal.
ADEM is typically a monophasic illness, and more than 90% of children experience only a single event. Some children may develop evolving or new neurologic deficits within 90 days of symptom onset; this is still considered part of the same ADEM event rather than a relapse. When new attacks occur more than 90 days after onset, they may represent multiphasic ADEM (MDEM) or another demyelinating syndrome such as optic neuritis or transverse myelitis. A more fulminant variant, acute hemorrhagic leukoencephalitis (AHLE), can resemble ADEM clinically but progresses more rapidly and is associated with multifocal hemorrhages on imaging.
Epidemiologically, ADEM is most prevalent in school-aged children, with a mean age of onset around 5 to 8 years, and a slight male predominance. Incidence is estimated at 0.1 to 0.6 per 100,000 per year, with peak occurrence in winter and spring. Because ADEM is generally transient, prevalence estimates are not well established. Although ADEM can occur in adults, adult-onset cases tend to have a poorer prognosis than pediatric cases.
Clinically, many patients have a febrile illness in the weeks before neurologic symptoms, often an upper respiratory infection. Children may present with headache, vomiting, vision changes, gait disturbance, meningismus, and seizures (reported in about one-third of patients). The hallmark feature is encephalopathy, ranging from irritability and confusion to lethargy and coma. The spinal cord may be involved (transverse myelitis), producing motor and sensory deficits and possible bowel or bladder dysfunction, and cranial neuropathies—including optic neuritis—may occur. The course can be rapidly progressive, and severe cases may require intensive care monitoring.
The etiology is strongly linked temporally to recent infections, and molecular mimicry has been proposed as a mechanism, though definitive proof is lacking. ADEM has been reported following various infections, and case reports have described ADEM after COVID-19 infection; however, a causal role for SARS-CoV-2 has not been established. Importantly, recent comprehensive studies have not shown associations between ADEM and common vaccines, and while rare reports exist after SARS-CoV-2 mRNA vaccination, no causal relationship has been demonstrated.
Diagnosis is clinical and requires encephalopathy plus polyfocal deficits; MRI abnormalities alone are not sufficient in the absence of the key clinical features. The differential diagnosis includes neuromyelitis optica spectrum disorder (NMOSD) associated with aquaporin-4 antibodies, MOG-associated disease (MOGAD), multiple sclerosis (MS), meningitis/encephalitis, brain mass lesions, CNS vasculitis, AHLE, hemophagocytic lymphohistiocytosis, and certain metabolic or mitochondrial disorders. Although ADEM can rarely be the first event of MS, MS criteria require evidence of dissemination in time and space (often supported by CSF oligoclonal bands) and typically a non-ADEM clinical pattern.
Workup generally includes evaluation for infectious meningoencephalitis when clinically indicated, cerebrospinal fluid (CSF) analysis, and targeted serum testing. CSF often shows pleocytosis reflecting inflammation. Oligoclonal bands that are present in CSF but not serum are uncommon in ADEM and, while nonspecific, are more typically associated with MS. Serum testing for MOG-IgG is important because approximately half of children with ADEM have MOG antibodies at presentation, and most cases of multiphasic ADEM are MOG-antibody positive. MOG testing should be performed in serum (live cell-based assays with clearly positive titers are preferred), because CSF testing is less specific for MOGAD. Testing for AQP4-IgG is used to identify NMOSD, which is usually a chronic relapsing disease with potentially poor recovery without appropriate long-term management.
Neuroimaging is central to evaluation. Head CT is often performed initially to exclude mass lesions but can miss demyelinating disease due to low sensitivity for white matter changes. Brain and spine MRI with and without contrast is recommended and commonly shows disseminated T2/FLAIR hyperintense lesions across supratentorial and infratentorial white matter, cortex, deep gray matter, brainstem, optic nerves, and sometimes the spinal cord. Lesions are often bilateral, large, poorly demarcated, and only a minority enhance with gadolinium. Follow-up MRI after monophasic ADEM typically demonstrates improvement or resolution over subsequent months.
Treatment is focused on rapid anti-inflammatory therapy and supportive care. Many patients—especially those with severe encephalopathy, seizures, brainstem involvement, or respiratory compromise—require ICU monitoring. First-line therapy is high-dose intravenous corticosteroids, commonly methylprednisolone 30 mg/kg/day (maximum 1000 mg/day) for 3 to 5 days. If there is inadequate clinical improvement, second-line therapies include intravenous immunoglobulin (IVIG) totaling 2 g/kg divided over 2 to 5 days, or 5 to 7 plasma exchanges.
Most patients improve with short-course corticosteroids and do not require a prolonged steroid taper. Long-term steroids are not indicated, even when MOG antibodies are present. Chronic immunomodulatory therapy is reserved for patients who are diagnosed with relapsing conditions such as NMOSD, relapsing MOGAD, or MS, and should be guided by neuroimmunology specialists. A single MOG-related demyelinating event generally does not require chronic immunotherapy, and currently there are no reliable biomarkers that predict which MOGAD patients will relapse.
Disposition depends on severity; significant encephalopathy, seizures, increased intracranial pressure concerns, or ventilatory dysfunction warrant inpatient—and often ICU—care until stabilization. Neurology consultation is recommended for diagnosis, treatment planning, and follow-up. Clinically, the appearance of relapses or new lesions beyond the expected monophasic window is not consistent with monophasic ADEM and should prompt reassessment for multiphasic ADEM, MOGAD, NMOSD, or MS.