- Published on
Emergency and Acute Medicine – Legg–Calvé–Perthes Disease
Basic description
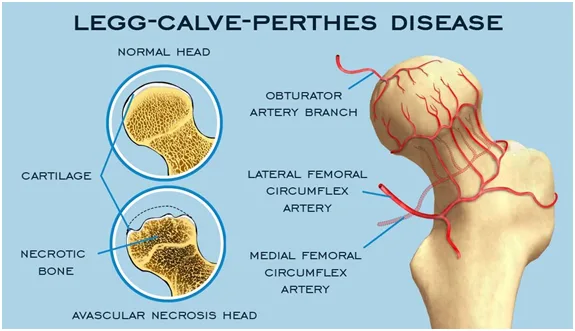
Legg–Calvé–Perthes disease is an idiopathic avascular necrosis of the femoral head occurring exclusively in children. It is thought to result from repeated vascular insults to the developing femoral head, leading to ischemia, collapse, and subsequent remodeling. Genetic associations include increased frequency in children with factor V Leiden mutation and anticardiolipin antibodies, supporting a possible hypercoagulable contribution.
Etiology
The condition is believed to arise from successive vascular occlusions of the femoral head and is likely multifactorial. Risk factors include male sex, Caucasian race, exposure to tobacco or wood smoke, low birth weight, and short birth length. The disease progresses through four stages: an initial ischemic stage with dense femoral head and synovitis, a fragmentation stage with femoral head softening and deformation, a healing stage with reossification, and a residual stage with variable permanent deformity. It most commonly affects children aged 3–7 years, with a male predominance of approximately 4:1, and is bilateral in 10–15% of cases.
Diagnosis: signs and symptoms
Presentation is often insidious. Limping is the most common initial complaint. Pain is typically dull or aching and may localize to the hip, groin, anteromedial thigh, or knee. Pain is activity related and improves with rest. Muscle spasm is common early in the disease course. On examination, children are usually afebrile and well appearing. Early findings include limited internal rotation and abduction of the hip. Later findings may include adductor contractures, muscle atrophy, and leg-length discrepancy. Some patients may be minimally symptomatic or asymptomatic.
Essential workup
Plain radiographs of the hips are the most important initial diagnostic study in the emergency setting. Septic arthritis must be considered and excluded, particularly in children with fever, acute onset, or toxic appearance.
Diagnostic tests and interpretation
No laboratory test is diagnostic for Legg–Calvé–Perthes disease. CBC, ESR, and CRP may be obtained if infection is a concern. Imaging establishes the diagnosis. AP and frog-leg lateral radiographs of both hips should be obtained to assess disease stage and detect bilateral involvement. Early radiographs may be normal in the first 3–6 months. MRI is more sensitive and can detect early ischemic changes and assess the extent of femoral head infarction. Bone scintigraphy can identify disease before radiographic changes but is less commonly used. Ultrasound may show a hip effusion but is nonspecific. CT and arthrography are primarily used for surgical planning rather than initial diagnosis.
Differential diagnosis
Unilateral disease considerations include transient synovitis, septic arthritis, osteomyelitis, sickle cell disease, juvenile idiopathic arthritis, trauma, slipped capital femoral epiphysis, tuberculosis, and malignancy. Bilateral disease raises consideration of hypothyroidism, epiphyseal dysplasia, and Gaucher disease.
Treatment
This is not a life-threatening condition, and emergency management focuses on symptom control. Pain management with NSAIDs is the primary intervention. Activity restriction is recommended, and crutches may be required if weight bearing is painful. Clinical instability, fever, or toxic appearance should prompt evaluation for alternative diagnoses.
Medications
First-line therapy is ibuprofen at 10 mg/kg per dose orally every 6–8 hours as needed for pain. Diazepam may be used as a second-line agent for significant muscle spasm.
Disposition and follow-up
Admission is rarely required and is reserved for severe pain not controlled with oral medications or social situations where home care is not feasible. Most patients can be discharged once pain is controlled, with orthopedic follow-up arranged within 1–2 weeks. Long-term management is determined by orthopedics and may include observation, activity modification, bracing, traction, or surgical intervention depending on age at onset and disease severity.
Pearls and pitfalls
Acute onset, fever, toxic appearance, or inability to bear weight should raise concern for diagnoses other than Legg–Calvé–Perthes disease, particularly septic arthritis. Early disease may have normal radiographs, so persistent symptoms warrant close follow-up and repeat imaging.
Basic description
Legg–Calvé–Perthes disease is an idiopathic avascular necrosis of the femoral head occurring exclusively in children. It is thought to result from repeated vascular insults to the developing femoral head, leading to ischemia, collapse, and subsequent remodeling. Genetic associations include increased frequency in children with factor V Leiden mutation and anticardiolipin antibodies, supporting a possible hypercoagulable contribution.
Etiology
The condition is believed to arise from successive vascular occlusions of the femoral head and is likely multifactorial. Risk factors include male sex, Caucasian race, exposure to tobacco or wood smoke, low birth weight, and short birth length. The disease progresses through four stages: an initial ischemic stage with dense femoral head and synovitis, a fragmentation stage with femoral head softening and deformation, a healing stage with reossification, and a residual stage with variable permanent deformity. It most commonly affects children aged 3–7 years, with a male predominance of approximately 4:1, and is bilateral in 10–15% of cases.
Diagnosis: signs and symptoms
Presentation is often insidious. Limping is the most common initial complaint. Pain is typically dull or aching and may localize to the hip, groin, anteromedial thigh, or knee. Pain is activity related and improves with rest. Muscle spasm is common early in the disease course. On examination, children are usually afebrile and well appearing. Early findings include limited internal rotation and abduction of the hip. Later findings may include adductor contractures, muscle atrophy, and leg-length discrepancy. Some patients may be minimally symptomatic or asymptomatic.
Essential workup
Plain radiographs of the hips are the most important initial diagnostic study in the emergency setting. Septic arthritis must be considered and excluded, particularly in children with fever, acute onset, or toxic appearance.
Diagnostic tests and interpretation
No laboratory test is diagnostic for Legg–Calvé–Perthes disease. CBC, ESR, and CRP may be obtained if infection is a concern. Imaging establishes the diagnosis. AP and frog-leg lateral radiographs of both hips should be obtained to assess disease stage and detect bilateral involvement. Early radiographs may be normal in the first 3–6 months. MRI is more sensitive and can detect early ischemic changes and assess the extent of femoral head infarction. Bone scintigraphy can identify disease before radiographic changes but is less commonly used. Ultrasound may show a hip effusion but is nonspecific. CT and arthrography are primarily used for surgical planning rather than initial diagnosis.
Differential diagnosis
Unilateral disease considerations include transient synovitis, septic arthritis, osteomyelitis, sickle cell disease, juvenile idiopathic arthritis, trauma, slipped capital femoral epiphysis, tuberculosis, and malignancy. Bilateral disease raises consideration of hypothyroidism, epiphyseal dysplasia, and Gaucher disease.
Treatment
This is not a life-threatening condition, and emergency management focuses on symptom control. Pain management with NSAIDs is the primary intervention. Activity restriction is recommended, and crutches may be required if weight bearing is painful. Clinical instability, fever, or toxic appearance should prompt evaluation for alternative diagnoses.
Medications
First-line therapy is ibuprofen at 10 mg/kg per dose orally every 6–8 hours as needed for pain. Diazepam may be used as a second-line agent for significant muscle spasm.
Disposition and follow-up
Admission is rarely required and is reserved for severe pain not controlled with oral medications or social situations where home care is not feasible. Most patients can be discharged once pain is controlled, with orthopedic follow-up arranged within 1–2 weeks. Long-term management is determined by orthopedics and may include observation, activity modification, bracing, traction, or surgical intervention depending on age at onset and disease severity.
Pearls and pitfalls
Acute onset, fever, toxic appearance, or inability to bear weight should raise concern for diagnoses other than Legg–Calvé–Perthes disease, particularly septic arthritis. Early disease may have normal radiographs, so persistent symptoms warrant close follow-up and repeat imaging.
- Published on
KembaraXtra- Medicine- Aphasia
Aphasia is an acquired disorder of language caused by brain damage to the dominant hemisphere (most often the left). The injury can result from vascular, traumatic, neurodegenerative, neoplastic, infectious, or inflammatory causes, with stroke being the most common. Aphasia is a language disorder, not a problem of muscle control or voice production, so it is different from dysarthria (neuromuscular speech control disorder), dysphonia (voice disorder), apraxia of speech (impaired programming of speech sounds), aphemia (muteness with preserved reading/writing/comprehension), and stuttering. Because language has a specific neuroanatomy, the symptoms of aphasia vary depending on which part of the language network is affected, including Broca area, Wernicke area, the arcuate fasciculus, and surrounding cortical regions.
Aphasia may be described using several related terms and syndromes, including expressive aphasia, receptive aphasia, conduction aphasia, global aphasia, and mixed transcortical (isolation) aphasia. Common ICD-10 codes include R47.01 (Aphasia) and several “aphasia following cerebrovascular disease” codes such as I69.320 (following cerebral infarction), I69.020 (following nontraumatic subarachnoid hemorrhage), I69.120 (following nontraumatic intracerebral hemorrhage), and I69.920 (following unspecified cerebrovascular disease). In the United States, more than one million stroke survivors live with stroke-related aphasia. Aphasia occurs in approximately 21% to 38% of acute stroke patients, and nearly 80% may still have aphasia at 12 months after stroke.
Clinically, aphasia is evaluated by determining whether speech is fluent or nonfluent and then systematically testing core language functions. Bedside assessment includes analysis of spontaneous speech, comprehension (following simple and complex commands), naming (common and uncommon objects), repetition (simple and complex phrases), and reading and writing. Aphasia can be obvious during conversation, but in acute settings it may be mistaken for—or occur alongside—encephalopathy or delirium, making careful language testing important. Stroke-related aphasia typically begins suddenly and is often accompanied by other neurologic signs consistent with involvement of the left middle cerebral artery territory or, in severe cases, the left internal carotid artery territory. Once the patient is stabilized and time-sensitive stroke treatments are addressed, a more detailed language examination is performed to classify the aphasia and guide localization and diagnosis.
Aphasias are broadly categorized into nonfluent and fluent types, with distinct patterns of comprehension and repetition. In nonfluent aphasias, speech is effortful and reduced. Broca aphasia features relatively intact comprehension but impaired repetition, while transcortical motor aphasia has intact comprehension and intact repetition. Global aphasia has impaired comprehension and repetition, and mixed transcortical (isolation) aphasia has impaired comprehension but preserved repetition. In fluent aphasias, speech output is more abundant and well-formed but may be meaningless or error-filled. Wernicke aphasia has impaired comprehension and impaired repetition, while transcortical sensory aphasia has impaired comprehension with preserved repetition. Conduction aphasia typically preserves comprehension but impairs repetition, and anomic aphasia preserves comprehension and repetition but has prominent word-finding difficulty. Salient differences include that nonfluent aphasia often presents with “telegraphic” speech (paucity of words, mainly nouns and verbs) and may be associated with right-sided weakness, whereas fluent aphasia commonly includes paraphasic errors, circumlocutions, tangential speech, and may be associated with visual field deficits or sensory loss, reflecting temporal or parietal involvement.
The differential diagnosis depends heavily on onset and tempo. Acute onset points strongly to stroke, but aphasia can also arise from seizures, trauma, tumors, infections, inflammatory disorders, or metabolic/toxic states affecting language networks. Progressive or insidious language decline suggests neurodegenerative disease, including Alzheimer disease (which can affect language early) and the primary progressive aphasias (PPAs), where language impairment is the dominant early feature. The major PPA variants include the nonfluent/agrammatic variant (nfvPPA), typically linked to degeneration of the left posterior frontoinsular region and often associated with frontotemporal lobar degeneration pathology; the semantic variant (svPPA), associated with anterior and ventral temporal lobe degeneration and characterized by impaired naming, impaired single-word comprehension, loss of object knowledge, and surface dyslexia; and the logopenic variant (lvPPA), linked to left posterior temporal and inferior parietal degeneration and marked by impaired word retrieval, impaired repetition of phrases and sentences, long pauses, and phonologic errors. Some svPPA cases may begin in the right anterior temporal lobe, initially affecting face recognition and interpretation of facial expressions before progressing to classic semantic language deficits.
Workup is targeted to the suspected cause but always includes neuroimaging, preferably brain MRI. In nonstroke aphasia (and when kidney function allows), MRI should be performed with and without contrast. In suspected acute stroke, urgent evaluation typically includes brain CT and CT angiography of the head and neck, with MRI often performed after acute interventions. Advanced imaging such as FDG-PET may be helpful in neurodegenerative aphasias to support diagnosis and characterize patterns of cortical dysfunction. Laboratory testing is likewise cause-directed: stroke-related aphasia evaluation includes assessment of vascular risk factors, while neurodegenerative aphasia workup parallels the broader assessment used for Alzheimer disease and related dementias.
Treatment focuses on the underlying etiology and on rehabilitation of language function. In the acute setting, management targets the cause (for example, stroke therapies when indicated). Long-term care includes speech-language therapy, using approaches such as script training, response elaboration training, constraint-induced aphasia therapy, speech entrainment, and melodic intonation therapy. Supportive communication strategies for patients and families are essential and include speaking slowly with natural rhythm, using short simple sentences, offering limited choices, asking yes/no questions, allowing extra time for responses, confirming understanding by repeating back key points, and avoiding over-correction of minor errors that do not change meaning. Environmental adjustments (reducing distractions, ensuring good lighting, facing the patient, keeping communication tools nearby) and visual supports (gestures, writing key words, pointing to objects or pictures, using communication books) can significantly improve day-to-day communication.
Referral commonly involves neurology for diagnosis and medical management, speech-language pathology for therapy, and often neuropsychology for cognitive assessment. Depending on associated needs, referrals may also include physical medicine and rehabilitation, and psychiatry, especially when mood, behavioral symptoms, or adjustment difficulties arise. Prognosis after stroke-related aphasia is difficult to predict, but recovery is influenced by lesion location and size, aphasia type and severity, and response to early interventions. In general, smaller lesions and preserved left superior temporal gyrus correlate with better outcomes, and intact basal ganglia may also support recovery. Broca and conduction aphasia often have better recovery trajectories than global aphasia, and recovery patterns vary widely across individuals.
Prevention of stroke-related aphasia centers on stroke prevention through modification of vascular risk factors. Education of patients and families should emphasize practical communication techniques and consistent supportive strategies to reduce frustration and improve participation in daily life.
Aphasia is an acquired disorder of language caused by brain damage to the dominant hemisphere (most often the left). The injury can result from vascular, traumatic, neurodegenerative, neoplastic, infectious, or inflammatory causes, with stroke being the most common. Aphasia is a language disorder, not a problem of muscle control or voice production, so it is different from dysarthria (neuromuscular speech control disorder), dysphonia (voice disorder), apraxia of speech (impaired programming of speech sounds), aphemia (muteness with preserved reading/writing/comprehension), and stuttering. Because language has a specific neuroanatomy, the symptoms of aphasia vary depending on which part of the language network is affected, including Broca area, Wernicke area, the arcuate fasciculus, and surrounding cortical regions.
Aphasia may be described using several related terms and syndromes, including expressive aphasia, receptive aphasia, conduction aphasia, global aphasia, and mixed transcortical (isolation) aphasia. Common ICD-10 codes include R47.01 (Aphasia) and several “aphasia following cerebrovascular disease” codes such as I69.320 (following cerebral infarction), I69.020 (following nontraumatic subarachnoid hemorrhage), I69.120 (following nontraumatic intracerebral hemorrhage), and I69.920 (following unspecified cerebrovascular disease). In the United States, more than one million stroke survivors live with stroke-related aphasia. Aphasia occurs in approximately 21% to 38% of acute stroke patients, and nearly 80% may still have aphasia at 12 months after stroke.
Clinically, aphasia is evaluated by determining whether speech is fluent or nonfluent and then systematically testing core language functions. Bedside assessment includes analysis of spontaneous speech, comprehension (following simple and complex commands), naming (common and uncommon objects), repetition (simple and complex phrases), and reading and writing. Aphasia can be obvious during conversation, but in acute settings it may be mistaken for—or occur alongside—encephalopathy or delirium, making careful language testing important. Stroke-related aphasia typically begins suddenly and is often accompanied by other neurologic signs consistent with involvement of the left middle cerebral artery territory or, in severe cases, the left internal carotid artery territory. Once the patient is stabilized and time-sensitive stroke treatments are addressed, a more detailed language examination is performed to classify the aphasia and guide localization and diagnosis.
Aphasias are broadly categorized into nonfluent and fluent types, with distinct patterns of comprehension and repetition. In nonfluent aphasias, speech is effortful and reduced. Broca aphasia features relatively intact comprehension but impaired repetition, while transcortical motor aphasia has intact comprehension and intact repetition. Global aphasia has impaired comprehension and repetition, and mixed transcortical (isolation) aphasia has impaired comprehension but preserved repetition. In fluent aphasias, speech output is more abundant and well-formed but may be meaningless or error-filled. Wernicke aphasia has impaired comprehension and impaired repetition, while transcortical sensory aphasia has impaired comprehension with preserved repetition. Conduction aphasia typically preserves comprehension but impairs repetition, and anomic aphasia preserves comprehension and repetition but has prominent word-finding difficulty. Salient differences include that nonfluent aphasia often presents with “telegraphic” speech (paucity of words, mainly nouns and verbs) and may be associated with right-sided weakness, whereas fluent aphasia commonly includes paraphasic errors, circumlocutions, tangential speech, and may be associated with visual field deficits or sensory loss, reflecting temporal or parietal involvement.
The differential diagnosis depends heavily on onset and tempo. Acute onset points strongly to stroke, but aphasia can also arise from seizures, trauma, tumors, infections, inflammatory disorders, or metabolic/toxic states affecting language networks. Progressive or insidious language decline suggests neurodegenerative disease, including Alzheimer disease (which can affect language early) and the primary progressive aphasias (PPAs), where language impairment is the dominant early feature. The major PPA variants include the nonfluent/agrammatic variant (nfvPPA), typically linked to degeneration of the left posterior frontoinsular region and often associated with frontotemporal lobar degeneration pathology; the semantic variant (svPPA), associated with anterior and ventral temporal lobe degeneration and characterized by impaired naming, impaired single-word comprehension, loss of object knowledge, and surface dyslexia; and the logopenic variant (lvPPA), linked to left posterior temporal and inferior parietal degeneration and marked by impaired word retrieval, impaired repetition of phrases and sentences, long pauses, and phonologic errors. Some svPPA cases may begin in the right anterior temporal lobe, initially affecting face recognition and interpretation of facial expressions before progressing to classic semantic language deficits.
Workup is targeted to the suspected cause but always includes neuroimaging, preferably brain MRI. In nonstroke aphasia (and when kidney function allows), MRI should be performed with and without contrast. In suspected acute stroke, urgent evaluation typically includes brain CT and CT angiography of the head and neck, with MRI often performed after acute interventions. Advanced imaging such as FDG-PET may be helpful in neurodegenerative aphasias to support diagnosis and characterize patterns of cortical dysfunction. Laboratory testing is likewise cause-directed: stroke-related aphasia evaluation includes assessment of vascular risk factors, while neurodegenerative aphasia workup parallels the broader assessment used for Alzheimer disease and related dementias.
Treatment focuses on the underlying etiology and on rehabilitation of language function. In the acute setting, management targets the cause (for example, stroke therapies when indicated). Long-term care includes speech-language therapy, using approaches such as script training, response elaboration training, constraint-induced aphasia therapy, speech entrainment, and melodic intonation therapy. Supportive communication strategies for patients and families are essential and include speaking slowly with natural rhythm, using short simple sentences, offering limited choices, asking yes/no questions, allowing extra time for responses, confirming understanding by repeating back key points, and avoiding over-correction of minor errors that do not change meaning. Environmental adjustments (reducing distractions, ensuring good lighting, facing the patient, keeping communication tools nearby) and visual supports (gestures, writing key words, pointing to objects or pictures, using communication books) can significantly improve day-to-day communication.
Referral commonly involves neurology for diagnosis and medical management, speech-language pathology for therapy, and often neuropsychology for cognitive assessment. Depending on associated needs, referrals may also include physical medicine and rehabilitation, and psychiatry, especially when mood, behavioral symptoms, or adjustment difficulties arise. Prognosis after stroke-related aphasia is difficult to predict, but recovery is influenced by lesion location and size, aphasia type and severity, and response to early interventions. In general, smaller lesions and preserved left superior temporal gyrus correlate with better outcomes, and intact basal ganglia may also support recovery. Broca and conduction aphasia often have better recovery trajectories than global aphasia, and recovery patterns vary widely across individuals.
Prevention of stroke-related aphasia centers on stroke prevention through modification of vascular risk factors. Education of patients and families should emphasize practical communication techniques and consistent supportive strategies to reduce frustration and improve participation in daily life.

- Published on
KembaraXtra- Medicine- Ascariasis
Ascariasis is a parasitic infection caused by the nematode Ascaris lumbricoides. Although most infected individuals remain asymptomatic, clinical disease can occur due to pulmonary hypersensitivity reactions, intestinal obstruction, nutrient depletion, or other secondary complications. Ascariasis remains the most common helminthic infection worldwide. The ICD-10-CM codes include B77.0 (with intestinal complications), B77.81 (ascariasis pneumonia), B77.89 (with other complications), and B77.9 (unspecified).
Globally, A. lumbricoides infects more than one billion people, with approximately 71% of those at risk living in Asia and the Western Pacific. In the United States, the exact incidence is unknown, but prevalence is estimated at about 4 million cases, primarily in rural areas of the southeastern states where sanitation is poor. Infection rates are reported to be approximately three times higher in Black populations than in White populations. Both sexes are affected equally, with a possible slight female predominance. Ascariasis is most common in children aged 2 to 10 years and tends to decrease after age 15. Infections often cluster within families. Neonatal infection is considered possible, although it has not been well studied.
Most individuals infected with Ascaris are asymptomatic. When symptoms occur, they often appear 9 to 12 days after ingestion of eggs, corresponding to larval migration through the lungs. Pulmonary manifestations include nonproductive cough, substernal chest discomfort, and fever, consistent with a hypersensitivity reaction. In patients with heavy worm burdens, particularly children, intestinal complications may occur, including obstruction, perforation, volvulus, and intussusception. Migration of adult worms into the biliary tree can produce symptoms resembling biliary colic, pancreatitis, or acute appendicitis. Rare complications include interstitial nephritis and acute renal failure. In endemic regions of Asia and Africa, chronic intestinal carriage may lead to malabsorption of dietary proteins and vitamins, contributing to malnutrition.
Transmission typically occurs via the fecal–oral route, most commonly through contaminated hands or ingestion of vegetables grown in soil containing infective eggs. After ingestion, eggs hatch in the small intestine, and larvae penetrate the intestinal mucosa, migrating via the bloodstream to the lungs. The larvae pass through the alveoli, ascend the bronchial tree, are swallowed, and return to the intestines, where they mature into adult worms. Female worms begin producing eggs approximately 2 to 3 months after infection. Eggs are excreted in feces and can survive for years in warm, moist, shaded soil. Adult worms live for approximately 1 to 2 years within the human host.
The differential diagnosis includes drug hypersensitivity reactions and Löffler syndrome when pulmonary infiltrates and eosinophilia are present. Other intestinal nematode infections should also be considered. Diagnosis is primarily established by laboratory evaluation, most commonly by identifying Ascaris ova in stool specimens. The World Health Organization recommends the Kato-Katz thick smear technique for detecting soil-transmitted helminths. Adult worms may also be passed in stool or expectorated. Eosinophilia is common early in infection, often ranging from 5% to 12%, but may reach as high as 50%, and typically decreases once adult worms are established in the intestines. Serologic testing detects IgG antibodies but lacks specificity due to cross-reactivity with other helminths and is mainly useful for epidemiologic purposes. Polymerase chain reaction testing of stool samples can identify A. lumbricoides and differentiate it from other parasites.
Imaging studies may support the diagnosis in complicated cases. Chest radiographs can demonstrate transient bilateral pulmonary infiltrates consistent with Löffler syndrome. Abdominal plain films or contrast studies may reveal worm masses within bowel loops. Ultrasonography and endoscopic retrograde cholangiopancreatography are useful for detecting worms in the pancreaticobiliary tract, while CT scanning with oral contrast can aid in identifying gastrointestinal obstruction caused by parasites.
All infected individuals, including those who are asymptomatic, should be treated. First-line therapy is albendazole 400 mg orally as a single dose. Mebendazole is an alternative, given as 100 mg orally twice daily for three days or as a single 500 mg dose where available. Cure rates with these agents are high, ranging from 95% to 100%, though both are classified as FDA pregnancy risk category C. The World Health Organization permits albendazole use during the second and third trimesters. Common side effects include gastrointestinal discomfort and headache, with rare cases of leukopenia. Pyrantel pamoate is a safe alternative during pregnancy and is administered at 11 mg/kg orally, with a maximum dose of 1 g. Other alternatives include ivermectin, nitazoxanide, piperazine citrate, and levamisole, though some are less effective or not available in the United States. Complete intestinal obstruction requires surgical management.
The overall prognosis of ascariasis is good, though reinfection is common. Patients should be reevaluated within 2 to 3 months after treatment. Referral to a gastroenterologist is indicated for pancreaticobiliary or appendiceal involvement, and surgical consultation is required for complete obstruction or complications such as perforation or volvulus.
Preventive measures are essential to reduce disease burden and include routine hand washing with soap, proper disposal of human waste, thorough washing or cooking of food, boiling drinking water, and preventing young children from direct contact with contaminated soil.
Ascariasis is a parasitic infection caused by the nematode Ascaris lumbricoides. Although most infected individuals remain asymptomatic, clinical disease can occur due to pulmonary hypersensitivity reactions, intestinal obstruction, nutrient depletion, or other secondary complications. Ascariasis remains the most common helminthic infection worldwide. The ICD-10-CM codes include B77.0 (with intestinal complications), B77.81 (ascariasis pneumonia), B77.89 (with other complications), and B77.9 (unspecified).
Globally, A. lumbricoides infects more than one billion people, with approximately 71% of those at risk living in Asia and the Western Pacific. In the United States, the exact incidence is unknown, but prevalence is estimated at about 4 million cases, primarily in rural areas of the southeastern states where sanitation is poor. Infection rates are reported to be approximately three times higher in Black populations than in White populations. Both sexes are affected equally, with a possible slight female predominance. Ascariasis is most common in children aged 2 to 10 years and tends to decrease after age 15. Infections often cluster within families. Neonatal infection is considered possible, although it has not been well studied.
Most individuals infected with Ascaris are asymptomatic. When symptoms occur, they often appear 9 to 12 days after ingestion of eggs, corresponding to larval migration through the lungs. Pulmonary manifestations include nonproductive cough, substernal chest discomfort, and fever, consistent with a hypersensitivity reaction. In patients with heavy worm burdens, particularly children, intestinal complications may occur, including obstruction, perforation, volvulus, and intussusception. Migration of adult worms into the biliary tree can produce symptoms resembling biliary colic, pancreatitis, or acute appendicitis. Rare complications include interstitial nephritis and acute renal failure. In endemic regions of Asia and Africa, chronic intestinal carriage may lead to malabsorption of dietary proteins and vitamins, contributing to malnutrition.
Transmission typically occurs via the fecal–oral route, most commonly through contaminated hands or ingestion of vegetables grown in soil containing infective eggs. After ingestion, eggs hatch in the small intestine, and larvae penetrate the intestinal mucosa, migrating via the bloodstream to the lungs. The larvae pass through the alveoli, ascend the bronchial tree, are swallowed, and return to the intestines, where they mature into adult worms. Female worms begin producing eggs approximately 2 to 3 months after infection. Eggs are excreted in feces and can survive for years in warm, moist, shaded soil. Adult worms live for approximately 1 to 2 years within the human host.
The differential diagnosis includes drug hypersensitivity reactions and Löffler syndrome when pulmonary infiltrates and eosinophilia are present. Other intestinal nematode infections should also be considered. Diagnosis is primarily established by laboratory evaluation, most commonly by identifying Ascaris ova in stool specimens. The World Health Organization recommends the Kato-Katz thick smear technique for detecting soil-transmitted helminths. Adult worms may also be passed in stool or expectorated. Eosinophilia is common early in infection, often ranging from 5% to 12%, but may reach as high as 50%, and typically decreases once adult worms are established in the intestines. Serologic testing detects IgG antibodies but lacks specificity due to cross-reactivity with other helminths and is mainly useful for epidemiologic purposes. Polymerase chain reaction testing of stool samples can identify A. lumbricoides and differentiate it from other parasites.
Imaging studies may support the diagnosis in complicated cases. Chest radiographs can demonstrate transient bilateral pulmonary infiltrates consistent with Löffler syndrome. Abdominal plain films or contrast studies may reveal worm masses within bowel loops. Ultrasonography and endoscopic retrograde cholangiopancreatography are useful for detecting worms in the pancreaticobiliary tract, while CT scanning with oral contrast can aid in identifying gastrointestinal obstruction caused by parasites.
All infected individuals, including those who are asymptomatic, should be treated. First-line therapy is albendazole 400 mg orally as a single dose. Mebendazole is an alternative, given as 100 mg orally twice daily for three days or as a single 500 mg dose where available. Cure rates with these agents are high, ranging from 95% to 100%, though both are classified as FDA pregnancy risk category C. The World Health Organization permits albendazole use during the second and third trimesters. Common side effects include gastrointestinal discomfort and headache, with rare cases of leukopenia. Pyrantel pamoate is a safe alternative during pregnancy and is administered at 11 mg/kg orally, with a maximum dose of 1 g. Other alternatives include ivermectin, nitazoxanide, piperazine citrate, and levamisole, though some are less effective or not available in the United States. Complete intestinal obstruction requires surgical management.
The overall prognosis of ascariasis is good, though reinfection is common. Patients should be reevaluated within 2 to 3 months after treatment. Referral to a gastroenterologist is indicated for pancreaticobiliary or appendiceal involvement, and surgical consultation is required for complete obstruction or complications such as perforation or volvulus.
Preventive measures are essential to reduce disease burden and include routine hand washing with soap, proper disposal of human waste, thorough washing or cooking of food, boiling drinking water, and preventing young children from direct contact with contaminated soil.
- Published on
KembaraXtra-Medicine – Astrocytoma
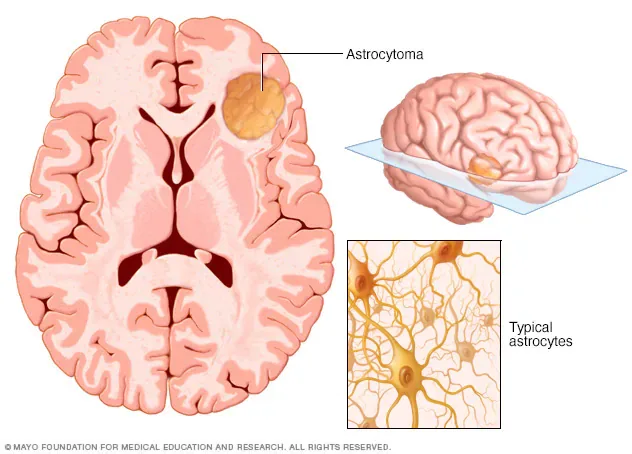
Astrocytomas are neuroepithelial tumors that arise from astrocytes, a type of glial precursor cell in the central nervous system. They are classified by the World Health Organization (WHO) into four grades based on histopathologic features, which provide important prognostic information. Grade I tumors are pilocytic astrocytomas and are generally benign. Grade II tumors are diffuse astrocytomas and are considered low grade. Grade III tumors are anaplastic astrocytomas, and Grade IV tumors are glioblastomas. Grades III and IV are classified as high-grade or malignant astrocytomas.
Astrocytomas account for approximately 10% of primary CNS tumors in the United States. In 2022, there were an estimated 25,050 new cases and 18,280 deaths related to primary CNS tumors, with an overall incidence of 6.4 cases per 100,000 persons per year. About half of malignant CNS tumors are glioblastomas. The clinical presentation of astrocytoma depends largely on tumor location and growth rate. Patients may present with headaches related to mass effect, new-onset partial or generalized seizures, nausea, vomiting, fatigue, and weakness. Focal neurologic deficits such as cranial nerve palsies, hemiplegia, ataxia, visual impairment, or language disturbances may occur. Cognitive or personality changes and, rarely, papilledema can also be seen.
The exact cause of astrocytomas remains unclear, although certain risk factors have been identified. Exposure to ionizing radiation is a known risk factor, and higher incidence has been observed in farmers and petrochemical workers. Several hereditary conditions are associated with increased risk, including neurofibromatosis type 1, Lynch syndrome, and Li-Fraumeni syndrome. At the molecular level, alterations in tumor suppressor genes such as TP53 play a significant role in astrocytoma development. Mutations in the IDH1 gene are common in grade II and III astrocytomas and are associated with a better prognosis compared with IDH-wild-type tumors. Molecular classification using TERT promoter mutations, IDH mutations, and 1p/19q codeletion further refines prognosis and treatment planning.
Diagnosis of astrocytoma is initially suspected based on clinical presentation and neuroimaging, but definitive diagnosis and grading require histopathologic confirmation. The differential diagnosis is broad and includes other causes of headaches, seizures, altered mental status, and focal neurologic deficits. Contrast-enhanced MRI is the imaging modality of choice because it provides detailed anatomic and pathologic information. CT scanning is reserved for patients unable to undergo MRI. Tissue diagnosis is obtained through surgical resection or stereotactic biopsy, particularly for deep-seated, multifocal, or unresectable tumors. Surgical goals include maximal safe tumor removal, reduction of mass effect and intracranial pressure, and procurement of tissue for diagnosis.
Management of astrocytoma is stage-specific and multidisciplinary. Corticosteroids such as dexamethasone are commonly used perioperatively to reduce cerebral edema. Maximal safe surgical resection is the cornerstone of treatment for all grades when feasible. Grade I astrocytomas are often cured with complete resection, while unresectable tumors may be observed until progression. Grade II astrocytomas benefit from surgery followed by adjuvant radiotherapy and chemotherapy, particularly when residual disease remains. Grade III anaplastic astrocytomas are treated with surgery followed by combined radiotherapy and chemotherapy, most commonly temozolomide. Grade IV glioblastomas require aggressive multimodal therapy, including maximal resection, concurrent chemoradiation with temozolomide, and adjuvant chemotherapy. Tumor-treating fields have shown additional survival benefit and are FDA-approved in selected patients.
Recurrent disease management depends on tumor grade and prior therapy. Options include repeat surgical resection, radiotherapy, chemotherapy, targeted therapies, and experimental approaches such as immunotherapy, tumor vaccines, and CAR-T cell therapy. Prognosis varies by grade, with excellent outcomes for grade I tumors, median survival of approximately 7.5 years for grade II, about 5 years for grade III, and roughly 14 months for glioblastoma. IDH-mutated tumors and those with favorable molecular features have improved survival.
Care of patients with astrocytoma requires coordinated management by neurosurgery, radiation oncology, and neuro-oncology teams to guide diagnosis, treatment, and long-term follow-up.
Astrocytomas are neuroepithelial tumors that arise from astrocytes, a type of glial precursor cell in the central nervous system. They are classified by the World Health Organization (WHO) into four grades based on histopathologic features, which provide important prognostic information. Grade I tumors are pilocytic astrocytomas and are generally benign. Grade II tumors are diffuse astrocytomas and are considered low grade. Grade III tumors are anaplastic astrocytomas, and Grade IV tumors are glioblastomas. Grades III and IV are classified as high-grade or malignant astrocytomas.
Astrocytomas account for approximately 10% of primary CNS tumors in the United States. In 2022, there were an estimated 25,050 new cases and 18,280 deaths related to primary CNS tumors, with an overall incidence of 6.4 cases per 100,000 persons per year. About half of malignant CNS tumors are glioblastomas. The clinical presentation of astrocytoma depends largely on tumor location and growth rate. Patients may present with headaches related to mass effect, new-onset partial or generalized seizures, nausea, vomiting, fatigue, and weakness. Focal neurologic deficits such as cranial nerve palsies, hemiplegia, ataxia, visual impairment, or language disturbances may occur. Cognitive or personality changes and, rarely, papilledema can also be seen.
The exact cause of astrocytomas remains unclear, although certain risk factors have been identified. Exposure to ionizing radiation is a known risk factor, and higher incidence has been observed in farmers and petrochemical workers. Several hereditary conditions are associated with increased risk, including neurofibromatosis type 1, Lynch syndrome, and Li-Fraumeni syndrome. At the molecular level, alterations in tumor suppressor genes such as TP53 play a significant role in astrocytoma development. Mutations in the IDH1 gene are common in grade II and III astrocytomas and are associated with a better prognosis compared with IDH-wild-type tumors. Molecular classification using TERT promoter mutations, IDH mutations, and 1p/19q codeletion further refines prognosis and treatment planning.
Diagnosis of astrocytoma is initially suspected based on clinical presentation and neuroimaging, but definitive diagnosis and grading require histopathologic confirmation. The differential diagnosis is broad and includes other causes of headaches, seizures, altered mental status, and focal neurologic deficits. Contrast-enhanced MRI is the imaging modality of choice because it provides detailed anatomic and pathologic information. CT scanning is reserved for patients unable to undergo MRI. Tissue diagnosis is obtained through surgical resection or stereotactic biopsy, particularly for deep-seated, multifocal, or unresectable tumors. Surgical goals include maximal safe tumor removal, reduction of mass effect and intracranial pressure, and procurement of tissue for diagnosis.
Management of astrocytoma is stage-specific and multidisciplinary. Corticosteroids such as dexamethasone are commonly used perioperatively to reduce cerebral edema. Maximal safe surgical resection is the cornerstone of treatment for all grades when feasible. Grade I astrocytomas are often cured with complete resection, while unresectable tumors may be observed until progression. Grade II astrocytomas benefit from surgery followed by adjuvant radiotherapy and chemotherapy, particularly when residual disease remains. Grade III anaplastic astrocytomas are treated with surgery followed by combined radiotherapy and chemotherapy, most commonly temozolomide. Grade IV glioblastomas require aggressive multimodal therapy, including maximal resection, concurrent chemoradiation with temozolomide, and adjuvant chemotherapy. Tumor-treating fields have shown additional survival benefit and are FDA-approved in selected patients.
Recurrent disease management depends on tumor grade and prior therapy. Options include repeat surgical resection, radiotherapy, chemotherapy, targeted therapies, and experimental approaches such as immunotherapy, tumor vaccines, and CAR-T cell therapy. Prognosis varies by grade, with excellent outcomes for grade I tumors, median survival of approximately 7.5 years for grade II, about 5 years for grade III, and roughly 14 months for glioblastoma. IDH-mutated tumors and those with favorable molecular features have improved survival.
Care of patients with astrocytoma requires coordinated management by neurosurgery, radiation oncology, and neuro-oncology teams to guide diagnosis, treatment, and long-term follow-up.
- Published on
KembaraXtra-Medicine – Asthma–COPD Overlap (ACO)
Asthma–COPD overlap (ACO) is a recognized clinical entity describing patients—often current or former smokers—who have chronic obstructive pulmonary disease (COPD) but also share important inflammatory and clinical features of asthma. Compared with COPD alone, ACO tends to be a more severe phenotype, with greater symptom burden and worse overall outcomes. ACO may be described as asthma with partially reversible airflow obstruction (with or without emphysema or reduced diffusing capacity) or as COPD with emphysema accompanied by reversible or partially reversible airflow obstruction (with or without allergies or reduced diffusing capacity). Because patients present with different patterns—such as COPD with eosinophilia, severe asthma in smokers, neutrophilic-predominant asthma, or asthma that has become largely irreversible due to airway remodeling—the term “ACO” is preferred over the older “ACOS” to reflect this heterogeneity.
ACO is discussed using overlapping ICD-10 codes including asthma (J45), chronic bronchitis (J42), emphysema (J43), COPD (J44, J44.9), and other overlap syndromes (M35.1). Epidemiologically, COPD prevalence varies across regions, and asthma affects tens of millions of people in the U.S. Bronchial hyperresponsiveness is common in COPD and bronchodilator reversibility can also be seen in COPD, which complicates diagnosis. Across studies, patients identified as having ACO generally demonstrate worse lung function, more respiratory symptoms, more frequent exacerbations, higher health-care utilization, and lower quality of life than those with asthma or COPD alone. Reported prevalence of ACO among adults with obstructive airway disease is commonly estimated around 15%–25%, with higher prevalence in older adults and in those with more severe disease. Some data suggest mortality in ACO is similar to COPD and worse than asthma alone.
Risk factors for ACO include cigarette smoking and atopy, along with older age, allergies, higher BMI, childhood asthma history, and respiratory infections such as rhinovirus or influenza. While no definitive genetic basis is established, some research has identified variants (including in GPR65) associated with ACO in certain populations. Clinically, patients may have wheezing, dyspnea, chest tightness, chronic cough (often productive), reduced exercise tolerance, recurrent respiratory infections, and episodic symptoms triggered by allergens, odors, or temperature changes. Physical examination can be normal or may show wheezing or rhonchi; in more severe cases, decreased air entry and accessory muscle use may appear, and signs such as the Hoover sign can suggest hyperinflation.
Diagnosis is considered when a patient demonstrates a mixed pattern of asthma-like and COPD-like features. A practical approach relies on objective confirmation of persistent airflow obstruction plus evidence of reversibility or airway hyperresponsiveness. Patients should have a reduced post-bronchodilator FEV₁/FVC (below the lower limit of normal or <0.7), along with either bronchodilator reversibility (increase in fev₁ or fvc by ≥200 ml and ≥12%) airway hyperresponsiveness demonstrated a positive methacholine challenge. because can occur copd symptoms overlap substantially, aco remains clinical diagnosis supported the pattern of symptoms, smoking history, asthma />llergy features, and pulmonary function testing rather than a single definitive test.
The differential diagnosis includes asthma, COPD, central airway obstruction, bronchiectasis, heart failure, and obliterative bronchiolitis. Workup typically includes spirometry with bronchodilator testing, and sometimes airway provocation testing. Additional supportive testing may include ABG (especially in severe disease), CBC with eosinophils, total IgE, sputum assessment, allergy testing, and peak flow monitoring. Imaging with chest x-ray or chest CT may help evaluate emphysema, alternative diagnoses, or complications, and ECG may be used when cardiac disease is suspected.
Management is challenging because many ACO patients have been excluded from major asthma and COPD drug trials, leaving limited high-quality evidence to guide therapy. A central reason to identify ACO is the potential for different responses to inhaled corticosteroids (ICS), particularly in patients with eosinophilic inflammation. Nonpharmacologic management includes smoking cessation, avoidance of triggers, inhaler technique education, and pulmonary rehabilitation; vaccination and oxygen supplementation may also be appropriate depending on severity. Pharmacologic treatment is generally symptom-directed, with bronchodilators used for dynamic obstruction and hyperinflation, while ICS is often included when asthma-like features or eosinophilia are present. Current guidance discourages using LABA and/or LAMA without ICS when asthma features are present, due to safety concerns and the need to treat underlying airway inflammation. Combination ICS/LABA therapy has evidence of benefit in some ACO populations, and observational data suggest improved outcomes compared with bronchodilator therapy alone in patients with overlapping asthma and COPD histories. Selected patients with allergic features may respond to biologic therapy such as omalizumab, with some observational data showing improvements similar to those seen in asthma without ACO.
Long-term care emphasizes earlier recognition in smokers or former smokers with partially reversible obstruction and progressive exercise intolerance, along with comprehensive management of comorbidities that worsen respiratory symptoms—such as GERD, chronic aspiration, vocal cord dysfunction, and nasal/sinus disease. Cardiovascular evaluation is especially important given higher cardiovascular risk in this population. Referral to a specialist is appropriate when symptoms or exacerbations persist, diagnosis remains uncertain, atypical features appear (e.g., hemoptysis, weight loss, fevers, night sweats, suspected bronchiectasis), comorbidities complicate management, or there is poor response to standard therapy. Prevention strategies focus on smoking cessation and minimizing exposure to triggers and irritants.
Asthma–COPD overlap (ACO) is a recognized clinical entity describing patients—often current or former smokers—who have chronic obstructive pulmonary disease (COPD) but also share important inflammatory and clinical features of asthma. Compared with COPD alone, ACO tends to be a more severe phenotype, with greater symptom burden and worse overall outcomes. ACO may be described as asthma with partially reversible airflow obstruction (with or without emphysema or reduced diffusing capacity) or as COPD with emphysema accompanied by reversible or partially reversible airflow obstruction (with or without allergies or reduced diffusing capacity). Because patients present with different patterns—such as COPD with eosinophilia, severe asthma in smokers, neutrophilic-predominant asthma, or asthma that has become largely irreversible due to airway remodeling—the term “ACO” is preferred over the older “ACOS” to reflect this heterogeneity.
ACO is discussed using overlapping ICD-10 codes including asthma (J45), chronic bronchitis (J42), emphysema (J43), COPD (J44, J44.9), and other overlap syndromes (M35.1). Epidemiologically, COPD prevalence varies across regions, and asthma affects tens of millions of people in the U.S. Bronchial hyperresponsiveness is common in COPD and bronchodilator reversibility can also be seen in COPD, which complicates diagnosis. Across studies, patients identified as having ACO generally demonstrate worse lung function, more respiratory symptoms, more frequent exacerbations, higher health-care utilization, and lower quality of life than those with asthma or COPD alone. Reported prevalence of ACO among adults with obstructive airway disease is commonly estimated around 15%–25%, with higher prevalence in older adults and in those with more severe disease. Some data suggest mortality in ACO is similar to COPD and worse than asthma alone.
Risk factors for ACO include cigarette smoking and atopy, along with older age, allergies, higher BMI, childhood asthma history, and respiratory infections such as rhinovirus or influenza. While no definitive genetic basis is established, some research has identified variants (including in GPR65) associated with ACO in certain populations. Clinically, patients may have wheezing, dyspnea, chest tightness, chronic cough (often productive), reduced exercise tolerance, recurrent respiratory infections, and episodic symptoms triggered by allergens, odors, or temperature changes. Physical examination can be normal or may show wheezing or rhonchi; in more severe cases, decreased air entry and accessory muscle use may appear, and signs such as the Hoover sign can suggest hyperinflation.
Diagnosis is considered when a patient demonstrates a mixed pattern of asthma-like and COPD-like features. A practical approach relies on objective confirmation of persistent airflow obstruction plus evidence of reversibility or airway hyperresponsiveness. Patients should have a reduced post-bronchodilator FEV₁/FVC (below the lower limit of normal or <0.7), along with either bronchodilator reversibility (increase in fev₁ or fvc by ≥200 ml and ≥12%) airway hyperresponsiveness demonstrated a positive methacholine challenge. because can occur copd symptoms overlap substantially, aco remains clinical diagnosis supported the pattern of symptoms, smoking history, asthma />llergy features, and pulmonary function testing rather than a single definitive test.
The differential diagnosis includes asthma, COPD, central airway obstruction, bronchiectasis, heart failure, and obliterative bronchiolitis. Workup typically includes spirometry with bronchodilator testing, and sometimes airway provocation testing. Additional supportive testing may include ABG (especially in severe disease), CBC with eosinophils, total IgE, sputum assessment, allergy testing, and peak flow monitoring. Imaging with chest x-ray or chest CT may help evaluate emphysema, alternative diagnoses, or complications, and ECG may be used when cardiac disease is suspected.
Management is challenging because many ACO patients have been excluded from major asthma and COPD drug trials, leaving limited high-quality evidence to guide therapy. A central reason to identify ACO is the potential for different responses to inhaled corticosteroids (ICS), particularly in patients with eosinophilic inflammation. Nonpharmacologic management includes smoking cessation, avoidance of triggers, inhaler technique education, and pulmonary rehabilitation; vaccination and oxygen supplementation may also be appropriate depending on severity. Pharmacologic treatment is generally symptom-directed, with bronchodilators used for dynamic obstruction and hyperinflation, while ICS is often included when asthma-like features or eosinophilia are present. Current guidance discourages using LABA and/or LAMA without ICS when asthma features are present, due to safety concerns and the need to treat underlying airway inflammation. Combination ICS/LABA therapy has evidence of benefit in some ACO populations, and observational data suggest improved outcomes compared with bronchodilator therapy alone in patients with overlapping asthma and COPD histories. Selected patients with allergic features may respond to biologic therapy such as omalizumab, with some observational data showing improvements similar to those seen in asthma without ACO.
Long-term care emphasizes earlier recognition in smokers or former smokers with partially reversible obstruction and progressive exercise intolerance, along with comprehensive management of comorbidities that worsen respiratory symptoms—such as GERD, chronic aspiration, vocal cord dysfunction, and nasal/sinus disease. Cardiovascular evaluation is especially important given higher cardiovascular risk in this population. Referral to a specialist is appropriate when symptoms or exacerbations persist, diagnosis remains uncertain, atypical features appear (e.g., hemoptysis, weight loss, fevers, night sweats, suspected bronchiectasis), comorbidities complicate management, or there is poor response to standard therapy. Prevention strategies focus on smoking cessation and minimizing exposure to triggers and irritants.
- Published on
KembaraXtra-Medicine – Asthma
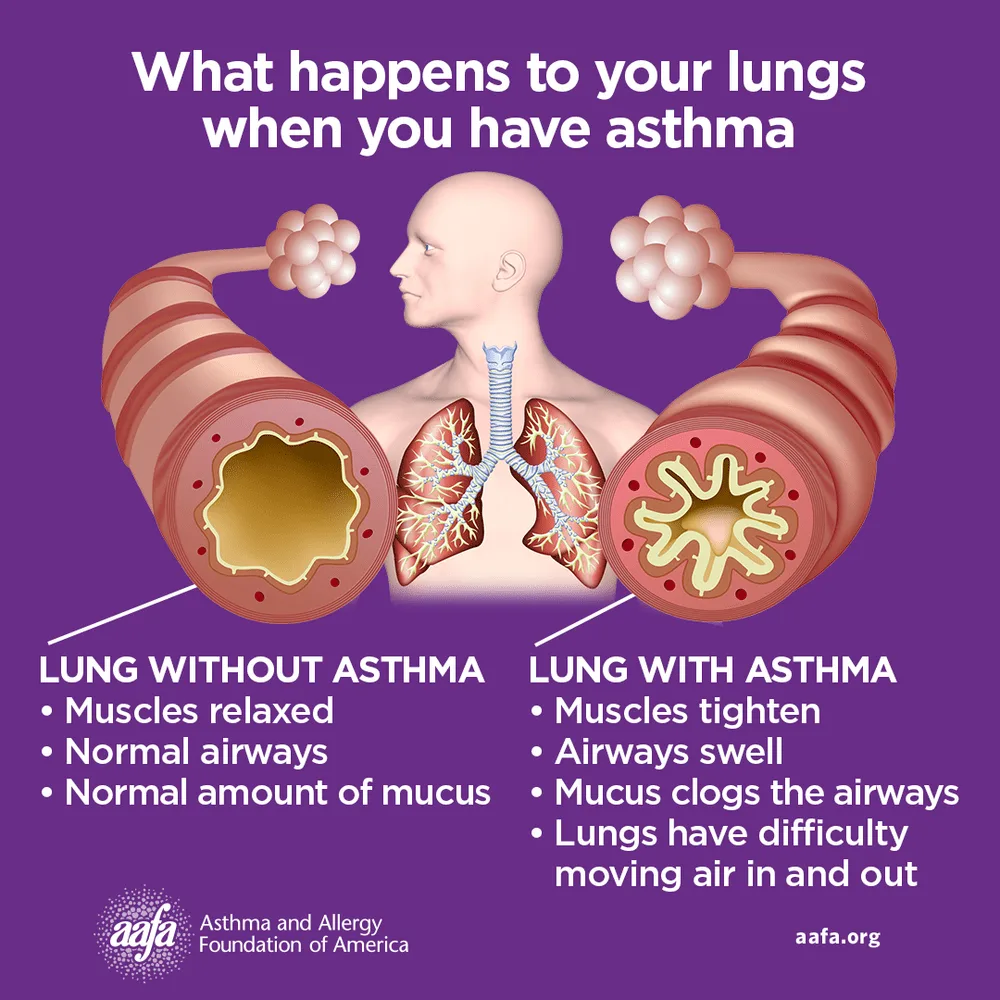
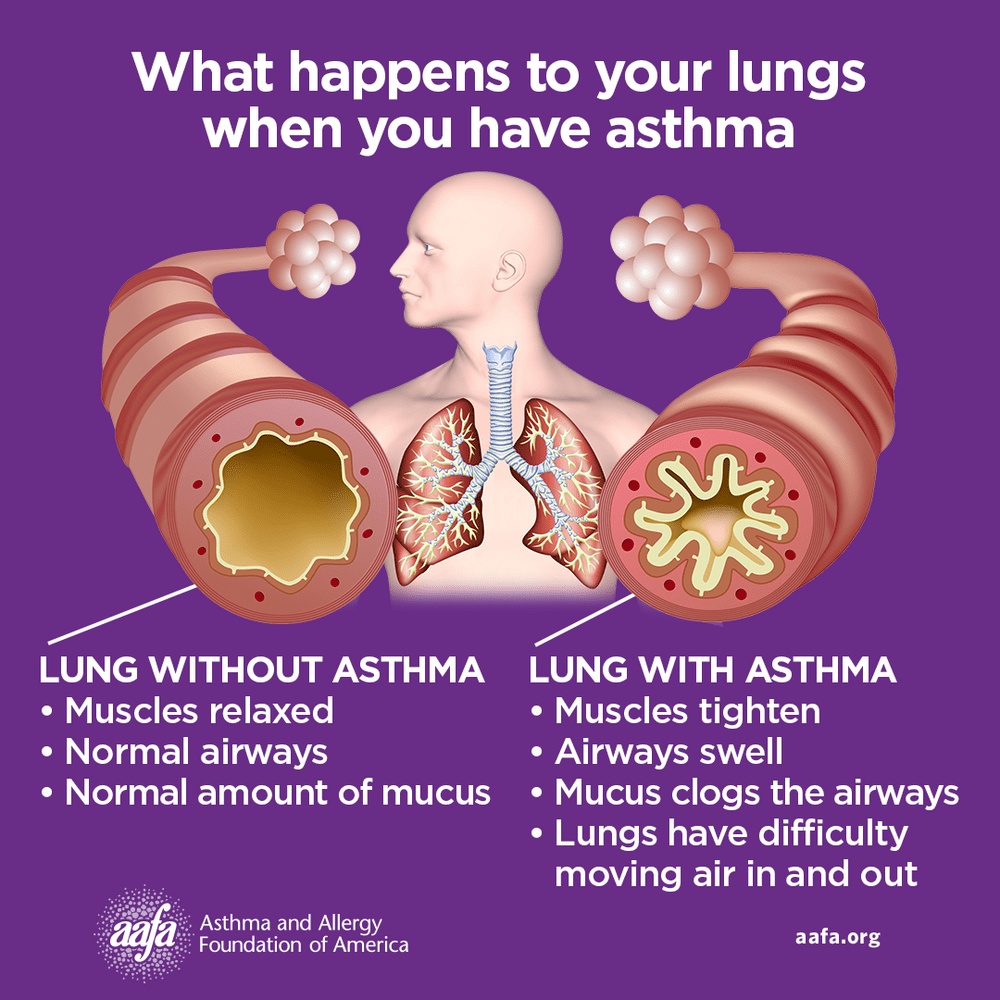
Asthma is a chronic inflammatory disease of the airways involving multiple cells, including mast cells, eosinophils, neutrophils, T lymphocytes, macrophages, and epithelial cells. This inflammation leads to recurrent episodes of cough (often at night or early morning), wheezing, shortness of breath, and chest tightness. These episodes are associated with variable and usually reversible airflow obstruction, either spontaneously or with treatment. Status asthmaticus refers to acute severe asthma that does not respond to standard therapies such as inhaled beta-agonists or subcutaneous epinephrine and may persist for hours, representing a life-threatening emergency.
Asthma is also referred to as bronchospasm, reactive airway disease, or asthmatic bronchitis. In the United States, approximately 7.7% of the population has asthma, with rising prevalence among adults, older individuals, women, African Americans, and people living below the poverty level. Globally, about 300 million people are affected, with projections reaching 400 million. Asthma accounts for significant healthcare use, including hundreds of thousands of hospitalizations and millions of emergency visits annually. Although mortality has declined overall, it remains unchanged in children aged 1–14 years. Many patients develop symptoms early in life, with 50–80% of children becoming symptomatic before age five.
Clinical presentation varies depending on severity and disease stage. Physical examination may be normal between attacks, but persistent or acute asthma often shows wheezing and prolonged expiration. Severe asthma and status asthmaticus may present with tachypnea, tachycardia, use of accessory muscles, pulsus paradoxus, altered mental status, paradoxical abdominal movement, or even a “silent chest,” which signals critical airflow obstruction. Risk factors for severe or fatal asthma include prior intubation, poor disease control, steroid dependence, smoking, obesity, psychiatric illness, advanced age, and limited access to medical care.
Asthma pathophysiology reflects a complex interaction between genetic susceptibility and environmental triggers. Allergic (atopic) asthma is driven by IgE-mediated responses to allergens, while nonallergic asthma often presents in adulthood following respiratory infections or stress. Occupational exposures, air pollutants, mold, exercise, medications such as NSAIDs or beta-blockers, and tobacco smoke can precipitate symptoms. Type-2 inflammatory pathways play a central role, involving cytokines such as IL-4, IL-5, and IL-13, leading to eosinophilia, IgE production, airway hyperresponsiveness, and remodeling. Genetic associations, including ADAM33 and other immune-related loci, further influence disease expression.
Diagnosis requires demonstration of airflow obstruction with reversibility. Spirometry before and after bronchodilator use is the preferred diagnostic test in patients older than five years, with reversibility defined as an increase in FEV₁ of at least 12% and 200 mL. Normal spirometry does not exclude asthma; bronchial challenge testing may be used when suspicion remains high. In young children, diagnosis is often clinical after exclusion of alternatives. Differential diagnoses include COPD, GERD, postnasal drip, vocal cord dysfunction, heart failure, pulmonary embolism, anxiety disorders, and interstitial lung disease.
Laboratory and ancillary testing help assess severity and guide management. Blood eosinophilia and elevated IgE support an allergic phenotype. Arterial blood gases are useful in acute severe attacks to assess hypoxia and hypercapnia. Imaging is usually normal but may show hyperinflation during exacerbations. Fractional exhaled nitric oxide and allergy testing can help phenotype asthma and predict response to therapy. Regular spirometry and peak flow monitoring are important for long-term assessment and self-management.
Management focuses on achieving and maintaining asthma control. Nonpharmacologic measures include trigger avoidance, smoking cessation, patient education, proper inhaler technique, and regular follow-up. Pharmacologic treatment follows a stepwise approach based on symptom frequency and severity. Short-acting beta-agonists are used for quick relief, while inhaled corticosteroids are the cornerstone of maintenance therapy. Additional controllers include long-acting beta-agonists (only in combination with inhaled corticosteroids), leukotriene receptor antagonists, long-acting muscarinic antagonists, and theophylline in selected cases. Therapy is stepped up when control is poor and stepped down when stable.
Patients with severe or refractory asthma may benefit from advanced therapies. Biologic agents targeting IgE, IL-5, IL-5 receptors, IL-4/IL-13 pathways, or thymic stromal lymphopoietin are used in carefully selected patients based on phenotype and biomarkers. Bronchial thermoplasty is an option for some adults with severe persistent asthma unresponsive to maximal medical therapy. Status asthmaticus requires aggressive management with oxygen, frequent bronchodilators, systemic corticosteroids, adjunctive agents such as magnesium sulfate, and ventilatory support when necessary.
Overall, asthma outcomes depend on individualized care, control of comorbid conditions, adherence to therapy, and early recognition of worsening symptoms. With appropriate management, most patients can achieve good symptom control, reduce exacerbations, and maintain normal activity levels.
Asthma is a chronic inflammatory disease of the airways involving multiple cells, including mast cells, eosinophils, neutrophils, T lymphocytes, macrophages, and epithelial cells. This inflammation leads to recurrent episodes of cough (often at night or early morning), wheezing, shortness of breath, and chest tightness. These episodes are associated with variable and usually reversible airflow obstruction, either spontaneously or with treatment. Status asthmaticus refers to acute severe asthma that does not respond to standard therapies such as inhaled beta-agonists or subcutaneous epinephrine and may persist for hours, representing a life-threatening emergency.
Asthma is also referred to as bronchospasm, reactive airway disease, or asthmatic bronchitis. In the United States, approximately 7.7% of the population has asthma, with rising prevalence among adults, older individuals, women, African Americans, and people living below the poverty level. Globally, about 300 million people are affected, with projections reaching 400 million. Asthma accounts for significant healthcare use, including hundreds of thousands of hospitalizations and millions of emergency visits annually. Although mortality has declined overall, it remains unchanged in children aged 1–14 years. Many patients develop symptoms early in life, with 50–80% of children becoming symptomatic before age five.
Clinical presentation varies depending on severity and disease stage. Physical examination may be normal between attacks, but persistent or acute asthma often shows wheezing and prolonged expiration. Severe asthma and status asthmaticus may present with tachypnea, tachycardia, use of accessory muscles, pulsus paradoxus, altered mental status, paradoxical abdominal movement, or even a “silent chest,” which signals critical airflow obstruction. Risk factors for severe or fatal asthma include prior intubation, poor disease control, steroid dependence, smoking, obesity, psychiatric illness, advanced age, and limited access to medical care.
Asthma pathophysiology reflects a complex interaction between genetic susceptibility and environmental triggers. Allergic (atopic) asthma is driven by IgE-mediated responses to allergens, while nonallergic asthma often presents in adulthood following respiratory infections or stress. Occupational exposures, air pollutants, mold, exercise, medications such as NSAIDs or beta-blockers, and tobacco smoke can precipitate symptoms. Type-2 inflammatory pathways play a central role, involving cytokines such as IL-4, IL-5, and IL-13, leading to eosinophilia, IgE production, airway hyperresponsiveness, and remodeling. Genetic associations, including ADAM33 and other immune-related loci, further influence disease expression.
Diagnosis requires demonstration of airflow obstruction with reversibility. Spirometry before and after bronchodilator use is the preferred diagnostic test in patients older than five years, with reversibility defined as an increase in FEV₁ of at least 12% and 200 mL. Normal spirometry does not exclude asthma; bronchial challenge testing may be used when suspicion remains high. In young children, diagnosis is often clinical after exclusion of alternatives. Differential diagnoses include COPD, GERD, postnasal drip, vocal cord dysfunction, heart failure, pulmonary embolism, anxiety disorders, and interstitial lung disease.
Laboratory and ancillary testing help assess severity and guide management. Blood eosinophilia and elevated IgE support an allergic phenotype. Arterial blood gases are useful in acute severe attacks to assess hypoxia and hypercapnia. Imaging is usually normal but may show hyperinflation during exacerbations. Fractional exhaled nitric oxide and allergy testing can help phenotype asthma and predict response to therapy. Regular spirometry and peak flow monitoring are important for long-term assessment and self-management.
Management focuses on achieving and maintaining asthma control. Nonpharmacologic measures include trigger avoidance, smoking cessation, patient education, proper inhaler technique, and regular follow-up. Pharmacologic treatment follows a stepwise approach based on symptom frequency and severity. Short-acting beta-agonists are used for quick relief, while inhaled corticosteroids are the cornerstone of maintenance therapy. Additional controllers include long-acting beta-agonists (only in combination with inhaled corticosteroids), leukotriene receptor antagonists, long-acting muscarinic antagonists, and theophylline in selected cases. Therapy is stepped up when control is poor and stepped down when stable.
Patients with severe or refractory asthma may benefit from advanced therapies. Biologic agents targeting IgE, IL-5, IL-5 receptors, IL-4/IL-13 pathways, or thymic stromal lymphopoietin are used in carefully selected patients based on phenotype and biomarkers. Bronchial thermoplasty is an option for some adults with severe persistent asthma unresponsive to maximal medical therapy. Status asthmaticus requires aggressive management with oxygen, frequent bronchodilators, systemic corticosteroids, adjunctive agents such as magnesium sulfate, and ventilatory support when necessary.
Overall, asthma outcomes depend on individualized care, control of comorbid conditions, adherence to therapy, and early recognition of worsening symptoms. With appropriate management, most patients can achieve good symptom control, reduce exacerbations, and maintain normal activity levels.
- Published on
KembaraXtra-Medicine – Aspergillosis
Aspergillosis is a group of illnesses caused by infection with Aspergillus species, a ubiquitous mold found worldwide in soil, building materials, and water. Humans are exposed mainly by inhaling airborne conidia (spores) that are small enough (about 2.5–3 μm) to reach the alveoli. Disease manifestations depend on the condition of the lungs, the host immune response, and the size of the inoculum. Aspergillus fumigatus is the most common human pathogen, while A. flavus is especially important in invasive disease and sinonasal involvement, and A. niger can also cause invasive infection. Hospital exposure can occur when unfiltered outside air enters through open windows or from contaminated water sources, and infections may also involve the nose, sinuses, external ear, or traumatized skin.
Risk is highest in immunocompromised patients, and invasive aspergillosis has become more common as modern therapies increasingly suppress immunity. Allogeneic stem cell transplant recipients and lung transplant recipients are among the highest-risk groups. A genetic deficiency of long pentraxin 3 (PTX3), which affects neutrophil antifungal function, may increase risk in hematopoietic stem-cell transplant patients. In advanced AIDS, patients with CD4 counts below 50/mm³ are more susceptible, although invasive aspergillosis remains otherwise uncommon in HIV. Severe influenza (including H1N1) can predispose to invasive disease by damaging respiratory epithelium, and COVID-19–associated pulmonary aspergillosis has been reported in a substantial proportion of patients with severe ARDS requiring intubation and is linked to higher mortality. Patients with chronic granulomatous disease are also at high risk, and A. nidulans is particularly associated with invasive infections in this group.
Pulmonary aspergillosis includes several clinical categories. Allergic fungal sinusitis occurs in patients with allergic rhinitis and is managed by sinus aeration and ensuring there is no tissue invasion, typically assessed by sinus CT. Allergic bronchopulmonary aspergillosis (ABPA) is a long-term allergic response seen most often in asthma and cystic fibrosis, driven by type I hypersensitivity and immune complex mechanisms, and it is commonly underdiagnosed. In ABPA, patients may develop worsening cough, wheeze, sputum production, and reduced pulmonary function; prevalence estimates range from about 1%–2% in persistently asthmatic patients and 2%–15% in cystic fibrosis. Chronic pulmonary aspergillosis is diagnosed when there are compatible thoracic imaging findings (preferably chest CT showing cavities with or without fungal ball or nodules), evidence of infection or immune response (such as elevated Aspergillus IgG/precipitins, strongly positive galactomannan or PCR), exclusion of alternative diagnoses, and symptoms or radiologic changes lasting at least 3 months.
Aspergillomas (“fungus balls”) occur when Aspergillus colonizes a preexisting lung cavity without tissue invasion or major immune response. These masses of hyphae usually develop in patients with underlying chronic lung disease such as prior tuberculosis, sarcoidosis, or emphysema. Many patients are asymptomatic, but hemoptysis is the most characteristic presentation, and imaging often shows an intracavitary mass with an air crescent that can move with patient position. Invasive aspergillosis is the most serious form and typically presents in immunocompromised patients as unremitting fever and new pulmonary infiltrates despite broad-spectrum antibiotics, along with dyspnea and nonproductive cough. Sudden pleuritic pain and tachycardia may mimic pulmonary embolism, and hemoptysis can occur. Imaging may show patchy bronchopneumonia, nodules, consolidation, cavitation, or characteristic CT findings such as the “halo sign” or “air-crescent sign.” In neutropenic hosts, incubation is often around 15 days. Invasive pulmonary aspergillosis is categorized as proven (sterile specimen with hyphae on microscopy or positive culture), probable (host factor plus clinical features plus mycologic evidence), or possible (host and clinical features without mycologic evidence).
Disseminated or extrapulmonary disease can occur in persistently immunosuppressed patients and may involve the brain, sinuses (with abscess formation by extension), gastrointestinal tract (ulcerations with risk of perforation or infarction), skin (necrotizing ulcers), bone (osteomyelitis), and even prosthetic or abnormal valves causing culture-negative endocarditis. Infections of implanted devices such as cardioverter-defibrillators have also been reported. Differential diagnoses vary by presentation and include tuberculosis, cystic fibrosis–related complications, lung carcinoma, eosinophilic pneumonia, bronchiectasis, sarcoidosis, and lung abscess.
Diagnosis depends on the clinical form. In ABPA, supportive laboratory features include peripheral eosinophilia, elevated total IgE, immediate skin test reactivity to Aspergillus, serum precipitins or IgG to A. fumigatus, and sometimes positive sputum cultures, though cultures are nonspecific. Diagnostic criteria commonly incorporate asthma or cystic fibrosis, evidence of immediate hypersensitivity or specific IgE, markedly elevated total IgE (often ≥1000 IU/mL, with some flexibility if other criteria are met), plus additional findings such as precipitins/IgG, radiographic opacities, or eosinophilia. In aspergilloma, sputum culture and serum precipitating antibodies may support the diagnosis, while imaging is often characteristic. In invasive aspergillosis, definitive diagnosis requires tissue invasion by septate, acute-angle branching hyphae or culture from tissue obtained via invasive sampling such as transbronchial biopsy. In high-risk patients, positive sputum or nasal cultures are strongly suggestive. Galactomannan antigen testing (Platelia Aspergillus ELISA) is widely used, especially in hematologic malignancy and stem-cell transplant populations; sensitivity may be reduced by ongoing antifungal therapy, and bronchoalveolar lavage galactomannan is often more sensitive than serum. Beta-D-glucan can detect early infection but is not specific for Aspergillus. Blood cultures are usually negative. PCR assays may be used in selected cases, but results should be interpreted alongside imaging, risk factors, and other mycologic data.
Imaging findings depend on the syndrome. ABPA may show fleeting patchy infiltrates, often in upper lobes, and many patients eventually develop central bronchiectasis. Aspergillomas are best seen on CXR or CT as an intracavity mass with an air crescent and may demonstrate movement with changes in patient positioning. In invasive disease, chest CT can reveal dense nodules that may cavitate, cavity formation, and the halo sign suggestive of angioinvasion, and CT is more sensitive than chest radiography in neutropenic patients.
Treatment is tailored to the specific form of aspergillosis. ABPA is treated with systemic corticosteroids such as prednisone (0.5–2 mg/kg/day orally) for 1–2 weeks followed by a slow taper over 2–3 months, combined with oral itraconazole (about 5 mg/kg/day up to 400 mg/day) with therapeutic drug monitoring for 3–6 months; voriconazole or posaconazole can be alternatives. Supportive care includes bronchodilators and physiotherapy, and serial chest imaging plus IgE levels can help track response. If prolonged steroids are needed, clinicians should consider prophylaxis against Pneumocystis jirovecii and strategies to preserve bone health.
Management of aspergilloma is challenging and sometimes controversial. Observation is reasonable for asymptomatic patients, and up to 10% may resolve without surgery or antifungals. For severe or life-threatening hemoptysis, surgical resection or arterial embolization is recommended. In patients at risk for major bleeding who have poor pulmonary reserve or are poor surgical candidates, antifungal therapy such as voriconazole may be considered, and in select cases intracavitary amphotericin B installation has been used. Resistance to itraconazole has increased in some Aspergillus strains, so local resistance patterns and prior azole exposure matter.
In invasive aspergillosis, first-line therapy is voriconazole (6 mg/kg IV/PO twice daily on day 1, then 4 mg/kg IV/PO twice daily), with therapeutic drug monitoring aiming for trough levels around 1.0–5.5 mg/L (often checked around day 4). Isavuconazonium sulfate (372 mg IV/PO three times daily for six doses, then 372 mg daily) and posaconazole (delayed-release tablets, suspension, or IV formulations with appropriate loading and maintenance regimens) are important alternatives, and posaconazole is also used for prophylaxis in selected high-risk groups such as neutropenic AML patients or those with graft-versus-host disease. Amphotericin B lipid complex or liposomal amphotericin B can be used as alternative therapy. Combination therapy may be considered in selected severe cases because azoles/polyenes target the cell membrane and echinocandins target the cell wall, but echinocandins should not be used as primary monotherapy for aspergillosis. Investigational agents such as olorofim and fosmanogepix are in clinical trials.
Special considerations include the high potential for drug–drug interactions with voriconazole, isavuconazole, and posaconazole, especially with chemotherapy and immunosuppressants, where azoles can raise drug levels and toxicity or alter efficacy. Azole resistance in A. fumigatus is an emerging concern, often related to CYP51A mutations; susceptibility testing should be considered in patients with substantial prior antifungal exposure or in areas with known high resistance rates. Hospital renovations and construction can increase airborne spores and infection risk in immunosuppressed patients. Coinfection with organisms such as Nocardia can occur, so persistent or atypical courses warrant broader evaluation. Referral to an infectious diseases specialist is recommended, particularly for invasive or disseminated disease, complex drug interactions, or refractory infections.
Aspergillosis is a group of illnesses caused by infection with Aspergillus species, a ubiquitous mold found worldwide in soil, building materials, and water. Humans are exposed mainly by inhaling airborne conidia (spores) that are small enough (about 2.5–3 μm) to reach the alveoli. Disease manifestations depend on the condition of the lungs, the host immune response, and the size of the inoculum. Aspergillus fumigatus is the most common human pathogen, while A. flavus is especially important in invasive disease and sinonasal involvement, and A. niger can also cause invasive infection. Hospital exposure can occur when unfiltered outside air enters through open windows or from contaminated water sources, and infections may also involve the nose, sinuses, external ear, or traumatized skin.
Risk is highest in immunocompromised patients, and invasive aspergillosis has become more common as modern therapies increasingly suppress immunity. Allogeneic stem cell transplant recipients and lung transplant recipients are among the highest-risk groups. A genetic deficiency of long pentraxin 3 (PTX3), which affects neutrophil antifungal function, may increase risk in hematopoietic stem-cell transplant patients. In advanced AIDS, patients with CD4 counts below 50/mm³ are more susceptible, although invasive aspergillosis remains otherwise uncommon in HIV. Severe influenza (including H1N1) can predispose to invasive disease by damaging respiratory epithelium, and COVID-19–associated pulmonary aspergillosis has been reported in a substantial proportion of patients with severe ARDS requiring intubation and is linked to higher mortality. Patients with chronic granulomatous disease are also at high risk, and A. nidulans is particularly associated with invasive infections in this group.
Pulmonary aspergillosis includes several clinical categories. Allergic fungal sinusitis occurs in patients with allergic rhinitis and is managed by sinus aeration and ensuring there is no tissue invasion, typically assessed by sinus CT. Allergic bronchopulmonary aspergillosis (ABPA) is a long-term allergic response seen most often in asthma and cystic fibrosis, driven by type I hypersensitivity and immune complex mechanisms, and it is commonly underdiagnosed. In ABPA, patients may develop worsening cough, wheeze, sputum production, and reduced pulmonary function; prevalence estimates range from about 1%–2% in persistently asthmatic patients and 2%–15% in cystic fibrosis. Chronic pulmonary aspergillosis is diagnosed when there are compatible thoracic imaging findings (preferably chest CT showing cavities with or without fungal ball or nodules), evidence of infection or immune response (such as elevated Aspergillus IgG/precipitins, strongly positive galactomannan or PCR), exclusion of alternative diagnoses, and symptoms or radiologic changes lasting at least 3 months.
Aspergillomas (“fungus balls”) occur when Aspergillus colonizes a preexisting lung cavity without tissue invasion or major immune response. These masses of hyphae usually develop in patients with underlying chronic lung disease such as prior tuberculosis, sarcoidosis, or emphysema. Many patients are asymptomatic, but hemoptysis is the most characteristic presentation, and imaging often shows an intracavitary mass with an air crescent that can move with patient position. Invasive aspergillosis is the most serious form and typically presents in immunocompromised patients as unremitting fever and new pulmonary infiltrates despite broad-spectrum antibiotics, along with dyspnea and nonproductive cough. Sudden pleuritic pain and tachycardia may mimic pulmonary embolism, and hemoptysis can occur. Imaging may show patchy bronchopneumonia, nodules, consolidation, cavitation, or characteristic CT findings such as the “halo sign” or “air-crescent sign.” In neutropenic hosts, incubation is often around 15 days. Invasive pulmonary aspergillosis is categorized as proven (sterile specimen with hyphae on microscopy or positive culture), probable (host factor plus clinical features plus mycologic evidence), or possible (host and clinical features without mycologic evidence).
Disseminated or extrapulmonary disease can occur in persistently immunosuppressed patients and may involve the brain, sinuses (with abscess formation by extension), gastrointestinal tract (ulcerations with risk of perforation or infarction), skin (necrotizing ulcers), bone (osteomyelitis), and even prosthetic or abnormal valves causing culture-negative endocarditis. Infections of implanted devices such as cardioverter-defibrillators have also been reported. Differential diagnoses vary by presentation and include tuberculosis, cystic fibrosis–related complications, lung carcinoma, eosinophilic pneumonia, bronchiectasis, sarcoidosis, and lung abscess.
Diagnosis depends on the clinical form. In ABPA, supportive laboratory features include peripheral eosinophilia, elevated total IgE, immediate skin test reactivity to Aspergillus, serum precipitins or IgG to A. fumigatus, and sometimes positive sputum cultures, though cultures are nonspecific. Diagnostic criteria commonly incorporate asthma or cystic fibrosis, evidence of immediate hypersensitivity or specific IgE, markedly elevated total IgE (often ≥1000 IU/mL, with some flexibility if other criteria are met), plus additional findings such as precipitins/IgG, radiographic opacities, or eosinophilia. In aspergilloma, sputum culture and serum precipitating antibodies may support the diagnosis, while imaging is often characteristic. In invasive aspergillosis, definitive diagnosis requires tissue invasion by septate, acute-angle branching hyphae or culture from tissue obtained via invasive sampling such as transbronchial biopsy. In high-risk patients, positive sputum or nasal cultures are strongly suggestive. Galactomannan antigen testing (Platelia Aspergillus ELISA) is widely used, especially in hematologic malignancy and stem-cell transplant populations; sensitivity may be reduced by ongoing antifungal therapy, and bronchoalveolar lavage galactomannan is often more sensitive than serum. Beta-D-glucan can detect early infection but is not specific for Aspergillus. Blood cultures are usually negative. PCR assays may be used in selected cases, but results should be interpreted alongside imaging, risk factors, and other mycologic data.
Imaging findings depend on the syndrome. ABPA may show fleeting patchy infiltrates, often in upper lobes, and many patients eventually develop central bronchiectasis. Aspergillomas are best seen on CXR or CT as an intracavity mass with an air crescent and may demonstrate movement with changes in patient positioning. In invasive disease, chest CT can reveal dense nodules that may cavitate, cavity formation, and the halo sign suggestive of angioinvasion, and CT is more sensitive than chest radiography in neutropenic patients.
Treatment is tailored to the specific form of aspergillosis. ABPA is treated with systemic corticosteroids such as prednisone (0.5–2 mg/kg/day orally) for 1–2 weeks followed by a slow taper over 2–3 months, combined with oral itraconazole (about 5 mg/kg/day up to 400 mg/day) with therapeutic drug monitoring for 3–6 months; voriconazole or posaconazole can be alternatives. Supportive care includes bronchodilators and physiotherapy, and serial chest imaging plus IgE levels can help track response. If prolonged steroids are needed, clinicians should consider prophylaxis against Pneumocystis jirovecii and strategies to preserve bone health.
Management of aspergilloma is challenging and sometimes controversial. Observation is reasonable for asymptomatic patients, and up to 10% may resolve without surgery or antifungals. For severe or life-threatening hemoptysis, surgical resection or arterial embolization is recommended. In patients at risk for major bleeding who have poor pulmonary reserve or are poor surgical candidates, antifungal therapy such as voriconazole may be considered, and in select cases intracavitary amphotericin B installation has been used. Resistance to itraconazole has increased in some Aspergillus strains, so local resistance patterns and prior azole exposure matter.
In invasive aspergillosis, first-line therapy is voriconazole (6 mg/kg IV/PO twice daily on day 1, then 4 mg/kg IV/PO twice daily), with therapeutic drug monitoring aiming for trough levels around 1.0–5.5 mg/L (often checked around day 4). Isavuconazonium sulfate (372 mg IV/PO three times daily for six doses, then 372 mg daily) and posaconazole (delayed-release tablets, suspension, or IV formulations with appropriate loading and maintenance regimens) are important alternatives, and posaconazole is also used for prophylaxis in selected high-risk groups such as neutropenic AML patients or those with graft-versus-host disease. Amphotericin B lipid complex or liposomal amphotericin B can be used as alternative therapy. Combination therapy may be considered in selected severe cases because azoles/polyenes target the cell membrane and echinocandins target the cell wall, but echinocandins should not be used as primary monotherapy for aspergillosis. Investigational agents such as olorofim and fosmanogepix are in clinical trials.
Special considerations include the high potential for drug–drug interactions with voriconazole, isavuconazole, and posaconazole, especially with chemotherapy and immunosuppressants, where azoles can raise drug levels and toxicity or alter efficacy. Azole resistance in A. fumigatus is an emerging concern, often related to CYP51A mutations; susceptibility testing should be considered in patients with substantial prior antifungal exposure or in areas with known high resistance rates. Hospital renovations and construction can increase airborne spores and infection risk in immunosuppressed patients. Coinfection with organisms such as Nocardia can occur, so persistent or atypical courses warrant broader evaluation. Referral to an infectious diseases specialist is recommended, particularly for invasive or disseminated disease, complex drug interactions, or refractory infections.
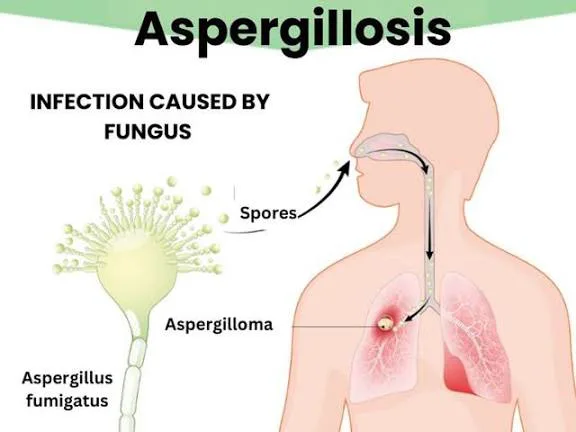
- Published on
KembaraXtra-Medicine – Asherman Syndrome (Intrauterine Adhesions)
Asherman syndrome refers to symptomatic intrauterine adhesions (IUA), which are bands of scar tissue, also known as synechiae, that form within the uterine cavity and sometimes the cervix. Synechiae develop when two opposing areas of endometrial tissue lose their epithelial lining and heal together, creating fibrous bridges. These adhesions may distort, narrow, or completely obliterate the uterine cavity. The term Asherman syndrome is reserved for cases in which intrauterine adhesions cause symptoms, whereas asymptomatic, incidentally discovered adhesions should be described simply as intrauterine adhesions.
The true incidence and prevalence of intrauterine adhesions in the general population are unknown because many cases are asymptomatic. Among individuals undergoing evaluation for infertility, approximately 1.5% are found to have intrauterine adhesions on hysterosalpingography, a figure that increases to over 20% in those with a prior history of dilation and curettage. Higher rates are also observed in women with recurrent miscarriage and in those who have undergone hysteroscopic uterine surgery, particularly repeat procedures. Asherman syndrome most commonly affects individuals of reproductive age, reflecting both exposure to intrauterine procedures and presentation with menstrual abnormalities or infertility.
Risk factors include dilation and curettage performed for miscarriage, abortion, abnormal uterine bleeding, or retained products of conception. Additional risks include placenta accreta, cesarean section, uterine compression sutures such as B-lynch sutures, abdominal or hysteroscopic myomectomy, pelvic inflammatory disease, genital tuberculosis, and surgical treatment of Müllerian anomalies. The risk of adhesion formation increases with repeated intrauterine procedures.
Many individuals with intrauterine adhesions are asymptomatic and never diagnosed. Symptomatic patients typically present with secondary amenorrhea, hypomenorrhea, infertility, or recurrent pregnancy loss. Some experience cyclic pelvic pain due to obstruction of menstrual flow. The severity of symptoms correlates with the extent and density of adhesions, which may range from thin, filmy bands to dense fibrotic tissue causing near-total obliteration of the uterine cavity. Adhesions are not palpable on bimanual examination and require imaging or direct visualization for diagnosis.
The pathophysiology of Asherman syndrome involves damage to the basal layer of the endometrium, which is responsible for regenerating the functional layer during the menstrual cycle. When the basal layer is destroyed, the endometrium cannot regenerate normally, leading to decreased or absent menstruation and impaired implantation. Inflammatory healing between opposing denuded uterine surfaces results in fibrous adhesion formation. The American Fertility Society classifies intrauterine adhesions as mild, moderate, or severe. Mild disease involves less than one-third of the uterine cavity and is associated with normal or mildly decreased menses. Moderate disease involves one- to two-thirds of the cavity with hypomenorrhea. Severe disease affects more than two-thirds of the cavity and typically presents with amenorrhea.
The differential diagnosis overlaps with that of secondary amenorrhea and infertility and includes cervical stenosis, thyroid disease, hypothalamic or pituitary dysfunction, premature ovarian insufficiency, polycystic ovarian syndrome, and androgen-secreting tumors. Intrauterine adhesions account for approximately 7% of cases of secondary amenorrhea.
Evaluation begins with laboratory assessment of ovarian and pituitary function, including follicle-stimulating hormone, luteinizing hormone, and estrogen levels. If these values are normal, a progesterone challenge test is performed. Failure to produce a withdrawal bleed suggests an outflow tract abnormality such as Asherman syndrome. Imaging is then pursued to evaluate the uterine cavity.
Saline-infusion sonography is the preferred screening modality and is more accurate than hysterosalpingography or transvaginal ultrasound alone. Adhesions appear as hypoechoic bands or filling defects within the uterine cavity. Hysterosalpingography may show compartmentalization or multiple filling defects. Definitive diagnosis is often made by hysteroscopy, which allows direct visualization of the adhesions.
Treatment is indicated for patients with symptomatic Asherman syndrome, including those with infertility, amenorrhea, hypomenorrhea, or cyclic pelvic pain. The primary goal is restoration of the normal uterine cavity through hysteroscopic adhesiolysis. Filmy adhesions may be divided with the hysteroscope alone, while denser adhesions require scissors or energy-based instruments. Adhesion division should proceed carefully from the center of the cavity outward to minimize further endometrial injury. There is no specific pharmacologic therapy for Asherman syndrome, although nonsteroidal anti-inflammatory drugs may be used for menstrual pain.
Prognosis depends on disease severity. Patients with mild adhesions generally have favorable outcomes, with improvement in menstrual function and fertility. Those with moderate to severe disease have lower fertility rates, even after surgical correction, as regeneration of the endometrial lining may be incomplete or delayed. Asymptomatic individuals who do not desire fertility do not require treatment.
Prevention focuses on minimizing unnecessary intrauterine procedures, particularly repeated dilation and curettage. When uterine evacuation is required, hysteroscopic techniques may reduce the risk of adhesion formation compared with blind curettage. Patients should be counseled about the risk of amenorrhea and infertility associated with intrauterine adhesions. Effective contraception can reduce the need for abortion-related procedures and thereby lower the risk of developing Asherman syndrome.
Asherman syndrome refers to symptomatic intrauterine adhesions (IUA), which are bands of scar tissue, also known as synechiae, that form within the uterine cavity and sometimes the cervix. Synechiae develop when two opposing areas of endometrial tissue lose their epithelial lining and heal together, creating fibrous bridges. These adhesions may distort, narrow, or completely obliterate the uterine cavity. The term Asherman syndrome is reserved for cases in which intrauterine adhesions cause symptoms, whereas asymptomatic, incidentally discovered adhesions should be described simply as intrauterine adhesions.
The true incidence and prevalence of intrauterine adhesions in the general population are unknown because many cases are asymptomatic. Among individuals undergoing evaluation for infertility, approximately 1.5% are found to have intrauterine adhesions on hysterosalpingography, a figure that increases to over 20% in those with a prior history of dilation and curettage. Higher rates are also observed in women with recurrent miscarriage and in those who have undergone hysteroscopic uterine surgery, particularly repeat procedures. Asherman syndrome most commonly affects individuals of reproductive age, reflecting both exposure to intrauterine procedures and presentation with menstrual abnormalities or infertility.
Risk factors include dilation and curettage performed for miscarriage, abortion, abnormal uterine bleeding, or retained products of conception. Additional risks include placenta accreta, cesarean section, uterine compression sutures such as B-lynch sutures, abdominal or hysteroscopic myomectomy, pelvic inflammatory disease, genital tuberculosis, and surgical treatment of Müllerian anomalies. The risk of adhesion formation increases with repeated intrauterine procedures.
Many individuals with intrauterine adhesions are asymptomatic and never diagnosed. Symptomatic patients typically present with secondary amenorrhea, hypomenorrhea, infertility, or recurrent pregnancy loss. Some experience cyclic pelvic pain due to obstruction of menstrual flow. The severity of symptoms correlates with the extent and density of adhesions, which may range from thin, filmy bands to dense fibrotic tissue causing near-total obliteration of the uterine cavity. Adhesions are not palpable on bimanual examination and require imaging or direct visualization for diagnosis.
The pathophysiology of Asherman syndrome involves damage to the basal layer of the endometrium, which is responsible for regenerating the functional layer during the menstrual cycle. When the basal layer is destroyed, the endometrium cannot regenerate normally, leading to decreased or absent menstruation and impaired implantation. Inflammatory healing between opposing denuded uterine surfaces results in fibrous adhesion formation. The American Fertility Society classifies intrauterine adhesions as mild, moderate, or severe. Mild disease involves less than one-third of the uterine cavity and is associated with normal or mildly decreased menses. Moderate disease involves one- to two-thirds of the cavity with hypomenorrhea. Severe disease affects more than two-thirds of the cavity and typically presents with amenorrhea.
The differential diagnosis overlaps with that of secondary amenorrhea and infertility and includes cervical stenosis, thyroid disease, hypothalamic or pituitary dysfunction, premature ovarian insufficiency, polycystic ovarian syndrome, and androgen-secreting tumors. Intrauterine adhesions account for approximately 7% of cases of secondary amenorrhea.
Evaluation begins with laboratory assessment of ovarian and pituitary function, including follicle-stimulating hormone, luteinizing hormone, and estrogen levels. If these values are normal, a progesterone challenge test is performed. Failure to produce a withdrawal bleed suggests an outflow tract abnormality such as Asherman syndrome. Imaging is then pursued to evaluate the uterine cavity.
Saline-infusion sonography is the preferred screening modality and is more accurate than hysterosalpingography or transvaginal ultrasound alone. Adhesions appear as hypoechoic bands or filling defects within the uterine cavity. Hysterosalpingography may show compartmentalization or multiple filling defects. Definitive diagnosis is often made by hysteroscopy, which allows direct visualization of the adhesions.
Treatment is indicated for patients with symptomatic Asherman syndrome, including those with infertility, amenorrhea, hypomenorrhea, or cyclic pelvic pain. The primary goal is restoration of the normal uterine cavity through hysteroscopic adhesiolysis. Filmy adhesions may be divided with the hysteroscope alone, while denser adhesions require scissors or energy-based instruments. Adhesion division should proceed carefully from the center of the cavity outward to minimize further endometrial injury. There is no specific pharmacologic therapy for Asherman syndrome, although nonsteroidal anti-inflammatory drugs may be used for menstrual pain.
Prognosis depends on disease severity. Patients with mild adhesions generally have favorable outcomes, with improvement in menstrual function and fertility. Those with moderate to severe disease have lower fertility rates, even after surgical correction, as regeneration of the endometrial lining may be incomplete or delayed. Asymptomatic individuals who do not desire fertility do not require treatment.
Prevention focuses on minimizing unnecessary intrauterine procedures, particularly repeated dilation and curettage. When uterine evacuation is required, hysteroscopic techniques may reduce the risk of adhesion formation compared with blind curettage. Patients should be counseled about the risk of amenorrhea and infertility associated with intrauterine adhesions. Effective contraception can reduce the need for abortion-related procedures and thereby lower the risk of developing Asherman syndrome.
- Published on
KembaraXtra- Medicine- Asbestosis
Asbestosis is a slowly progressive, diffuse interstitial pulmonary fibrosis caused by dose-related inhalation of asbestos fibers. It most commonly affects miners, millers, asbestos textile workers, and insulators. Clinically, lung involvement is characterized by bilateral diffuse interstitial fibrosis that is more pronounced in the lower lobes, along with pleural thickening, leading to progressive shortness of breath and a dry cough. Asbestos exposure can result in a spectrum of pulmonary pathology, including pulmonary fibrosis, asbestos-related pleural plaque disease (both focal and diffuse), and malignancies such as lung cancer and mesothelioma. The ICD-10-CM code for asbestosis is J61.
The incidence of asbestosis in the United States is approximately 5 to 10 new cases per 100,000 persons per year. There is typically a prolonged latency period of 20 to 30 years between asbestos exposure and the onset of clinical disease. The condition is most commonly seen in individuals over 40 years of age who were involved in asbestos extraction or in the manufacture and installation of asbestos-containing materials, including those working in naval shipyards during World War II and in construction settings involving insulation, tiles, and pipe coverings. Smokers and heavy alcohol users are at particularly high risk.
Clinically, asbestosis presents insidiously, with exertional dyspnea and a dry, nonproductive cough often being the earliest symptoms. As the disease progresses, dyspnea worsens and patients tolerate progressively less physical exertion. The cough is typically paroxysmal and dry, though scant mucoid sputum may appear in advanced stages. Hemoptysis is rare. Physical examination often reveals fine end-inspiratory crackles, especially at the lung bases. Digital clubbing, peripheral edema, jugular venous distention, and signs of right-sided heart failure may be present in advanced disease.
The pathogenesis of asbestosis is related to inhalation of asbestos fibers, which induce immune-mediated pulmonary interstitial inflammation and fibrosis. Asbestosis has been associated with positive serum antinuclear antibodies and rheumatoid factor, suggesting an autoimmune component. Interleukin-1 beta (IL-1β) plays an important role in disease progression and in associated systemic autoimmune manifestations.
Asbestos exposure can lead to several pulmonary manifestations. Pleural plaques are the most common and are usually asymptomatic radiographic findings appearing 20 to 30 years after exposure. Diffuse pleural thickening involves the visceral pleura and is thought to result from chronic pleural irritation; it usually does not cause symptoms. Rounded atelectasis occurs adjacent to the pleura and shows the characteristic “comet tail” sign on imaging; it is typically asymptomatic but warrants evaluation if associated with pleural effusion. Benign asbestos-related pleural effusions are usually unilateral, exudative, and eosinophil-predominant, often resolving spontaneously but sometimes recurring. Asbestos-related interstitial lung disease ranges from mild fibrosis to progressive diffuse pulmonary fibrosis and must be distinguished from other interstitial lung diseases such as usual interstitial pneumonia. Asbestos exposure is also strongly associated with malignancy, particularly bronchogenic carcinoma and mesothelioma, with risk markedly increased in smokers.
The differential diagnosis of asbestosis includes silicosis, siderosis and other pneumoconioses, interstitial lung disease, and lung cancer. Diagnostic evaluation relies on careful documentation of asbestos exposure history, pulmonary function testing, and imaging studies. Laboratory tests are generally not helpful, though arterial blood gases may demonstrate hypoxemia or hypercarbia in advanced disease.
Pulmonary function tests typically reveal a restrictive pattern with reduced vital capacity, total lung capacity, and diffusing capacity for carbon monoxide (DLco). Forced expiratory volume in one second (FEV₁) may also be reduced in smokers with concomitant obstructive disease. Bronchoscopy with bronchoalveolar lavage has limited diagnostic utility but may help exclude infection or hypersensitivity pneumonitis; asbestos bodies may be identified but are not diagnostic of disease severity or progression.
Imaging studies are central to diagnosis. Chest radiographs may show pleural plaques, pleural calcifications, diffuse pleural thickening, blunting of costophrenic angles, interlobar fissure thickening, or diffuse interstitial fibrosis. High-resolution computed tomography of the chest is more sensitive and typically demonstrates basilar-predominant interstitial markings, with honeycombing seen in advanced disease.
There is no specific pharmacologic therapy for asbestosis. Management focuses on supportive and preventive measures. Nonpharmacologic treatment includes smoking cessation, avoidance of further asbestos exposure, regular exercise or pulmonary rehabilitation to optimize functional capacity, supplemental oxygen as needed, and prompt treatment of respiratory infections. Vaccination against influenza and pneumococcal disease is recommended. Emerging data suggest that therapies targeting IL-1β may have future potential, particularly for systemic autoimmune features, though no definitive treatment is currently available.
The prognosis of asbestosis depends on disease severity and complications. Death most commonly results from respiratory failure due to cor pulmonale. Survival after the development of mesothelioma is typically 4 to 6 years. Benign asbestos pleural effusions often precede interstitial disease by several years. Diffuse pleural thickening and asbestosis are associated with an increased risk of malignant peritoneal mesothelioma, although benign pleural disease alone does not increase the risk of malignant pleural mesothelioma. Asbestos exposure significantly increases lung cancer risk regardless of smoking status, with a synergistic effect in smokers. Patients with asbestosis who smoke have up to a 40-fold increased risk of lung cancer compared with never-smokers. Smoking cessation dramatically reduces this risk over time. Low-dose chest CT screening is valuable for early detection of lung cancer in asbestos-exposed individuals, and CT imaging is generally sufficient for evaluation, with PET scanning reserved for suspected mesothelioma.
Asbestosis is a slowly progressive, diffuse interstitial pulmonary fibrosis caused by dose-related inhalation of asbestos fibers. It most commonly affects miners, millers, asbestos textile workers, and insulators. Clinically, lung involvement is characterized by bilateral diffuse interstitial fibrosis that is more pronounced in the lower lobes, along with pleural thickening, leading to progressive shortness of breath and a dry cough. Asbestos exposure can result in a spectrum of pulmonary pathology, including pulmonary fibrosis, asbestos-related pleural plaque disease (both focal and diffuse), and malignancies such as lung cancer and mesothelioma. The ICD-10-CM code for asbestosis is J61.
The incidence of asbestosis in the United States is approximately 5 to 10 new cases per 100,000 persons per year. There is typically a prolonged latency period of 20 to 30 years between asbestos exposure and the onset of clinical disease. The condition is most commonly seen in individuals over 40 years of age who were involved in asbestos extraction or in the manufacture and installation of asbestos-containing materials, including those working in naval shipyards during World War II and in construction settings involving insulation, tiles, and pipe coverings. Smokers and heavy alcohol users are at particularly high risk.
Clinically, asbestosis presents insidiously, with exertional dyspnea and a dry, nonproductive cough often being the earliest symptoms. As the disease progresses, dyspnea worsens and patients tolerate progressively less physical exertion. The cough is typically paroxysmal and dry, though scant mucoid sputum may appear in advanced stages. Hemoptysis is rare. Physical examination often reveals fine end-inspiratory crackles, especially at the lung bases. Digital clubbing, peripheral edema, jugular venous distention, and signs of right-sided heart failure may be present in advanced disease.
The pathogenesis of asbestosis is related to inhalation of asbestos fibers, which induce immune-mediated pulmonary interstitial inflammation and fibrosis. Asbestosis has been associated with positive serum antinuclear antibodies and rheumatoid factor, suggesting an autoimmune component. Interleukin-1 beta (IL-1β) plays an important role in disease progression and in associated systemic autoimmune manifestations.
Asbestos exposure can lead to several pulmonary manifestations. Pleural plaques are the most common and are usually asymptomatic radiographic findings appearing 20 to 30 years after exposure. Diffuse pleural thickening involves the visceral pleura and is thought to result from chronic pleural irritation; it usually does not cause symptoms. Rounded atelectasis occurs adjacent to the pleura and shows the characteristic “comet tail” sign on imaging; it is typically asymptomatic but warrants evaluation if associated with pleural effusion. Benign asbestos-related pleural effusions are usually unilateral, exudative, and eosinophil-predominant, often resolving spontaneously but sometimes recurring. Asbestos-related interstitial lung disease ranges from mild fibrosis to progressive diffuse pulmonary fibrosis and must be distinguished from other interstitial lung diseases such as usual interstitial pneumonia. Asbestos exposure is also strongly associated with malignancy, particularly bronchogenic carcinoma and mesothelioma, with risk markedly increased in smokers.
The differential diagnosis of asbestosis includes silicosis, siderosis and other pneumoconioses, interstitial lung disease, and lung cancer. Diagnostic evaluation relies on careful documentation of asbestos exposure history, pulmonary function testing, and imaging studies. Laboratory tests are generally not helpful, though arterial blood gases may demonstrate hypoxemia or hypercarbia in advanced disease.
Pulmonary function tests typically reveal a restrictive pattern with reduced vital capacity, total lung capacity, and diffusing capacity for carbon monoxide (DLco). Forced expiratory volume in one second (FEV₁) may also be reduced in smokers with concomitant obstructive disease. Bronchoscopy with bronchoalveolar lavage has limited diagnostic utility but may help exclude infection or hypersensitivity pneumonitis; asbestos bodies may be identified but are not diagnostic of disease severity or progression.
Imaging studies are central to diagnosis. Chest radiographs may show pleural plaques, pleural calcifications, diffuse pleural thickening, blunting of costophrenic angles, interlobar fissure thickening, or diffuse interstitial fibrosis. High-resolution computed tomography of the chest is more sensitive and typically demonstrates basilar-predominant interstitial markings, with honeycombing seen in advanced disease.
There is no specific pharmacologic therapy for asbestosis. Management focuses on supportive and preventive measures. Nonpharmacologic treatment includes smoking cessation, avoidance of further asbestos exposure, regular exercise or pulmonary rehabilitation to optimize functional capacity, supplemental oxygen as needed, and prompt treatment of respiratory infections. Vaccination against influenza and pneumococcal disease is recommended. Emerging data suggest that therapies targeting IL-1β may have future potential, particularly for systemic autoimmune features, though no definitive treatment is currently available.
The prognosis of asbestosis depends on disease severity and complications. Death most commonly results from respiratory failure due to cor pulmonale. Survival after the development of mesothelioma is typically 4 to 6 years. Benign asbestos pleural effusions often precede interstitial disease by several years. Diffuse pleural thickening and asbestosis are associated with an increased risk of malignant peritoneal mesothelioma, although benign pleural disease alone does not increase the risk of malignant pleural mesothelioma. Asbestos exposure significantly increases lung cancer risk regardless of smoking status, with a synergistic effect in smokers. Patients with asbestosis who smoke have up to a 40-fold increased risk of lung cancer compared with never-smokers. Smoking cessation dramatically reduces this risk over time. Low-dose chest CT screening is valuable for early detection of lung cancer in asbestos-exposed individuals, and CT imaging is generally sufficient for evaluation, with PET scanning reserved for suspected mesothelioma.
- Published on
KembaraXtra- Medicine- Aphthous Ulcers
Aphthous ulcers are small, painful ulcers that occur predominantly on the oral mucosa. They are also known as aphthae, canker sores, recurrent oral aphthae, or recurrent aphthous stomatitis (RAS). The ICD-10CM code is K12.0 (Recurrent oral aphthae). The incidence is unknown, but prevalence is common—about one in five people experience them at some point in life. They most commonly affect women, adolescents, and adults under 40, with peak incidence in adolescence. Risk factors include genetic predisposition, stress, local trauma, tobacco exposure, viral or bacterial infections, nutritional deficiencies, and endocrine or autoimmune diseases. Genetics play a role, as up to 40% of patients report a family history.
Clinically, aphthous ulcers are typically small round or oval lesions with a gray-white or yellow pseudomembranous base and an erythematous (red) halo. They can occur at any age, often beginning in childhood and commonly improving by the third decade. The exact cause is not well understood and is likely multifactorial, with contributors such as trauma (biting, dental appliances/braces), nutritional deficiencies, infections, medications, food hypersensitivity, hormonal/endocrine changes, and tobacco use. When ulcers recur without an identified systemic disease, the condition is termed recurrent aphthous stomatitis (RAS).
RAS is divided into three subtypes. Minor RAS (about 80%) presents as 1 to 5 shallow ulcers that are <10 mm, usually on the lips, buccal mucosa, tongue, and floor of mouth, they typically resolve in 4 to 14 days without scarring. major ras (about 10%) features larger (>10 mm), deeper ulcers that may be more numerous and extend to areas like the gingiva and pharyngeal mucosa; they last weeks to months and have a higher risk of scarring. Herpetiform RAS (about 10%) consists of many very painful small ulcers (1–3 mm) occurring in large numbers (5 to 100), often involving the floor of mouth and ventral tongue; ulcers may cluster and coalesce into larger irregular lesions, generally resolving within 14 days and usually without visible scarring. Despite the name, herpetiform RAS is not caused by herpes.
The differential diagnosis is broad, especially when ulcers are severe, persistent, atypical, or associated with systemic findings. Important considerations include Behçet syndrome, MAGIC syndrome, systemic lupus erythematosus, gluten-sensitive enteropathy, inflammatory bowel disease, HIV infection, herpes simplex virus, cyclic neutropenia, PFAPA syndrome, hyperimmunoglobulin D syndrome, agranulocytosis, autoimmune bullous disease, oral erosive lichen planus, drug-induced mucosal ulcers, contact dermatitis, and squamous cell carcinoma. Differentiating HSV from aphthous ulcers is useful: HSV tends to involve keratinized tissue, often has a vesicle phase and prodrome, whereas aphthous ulcers usually occur on movable, nonkeratinized mucosa, lack vesicles, and have nonspecific biopsy findings if biopsied.
Workup begins with a detailed clinical history (associated symptoms, family history, frequency, number/size, duration, location, and presence of any nonoral lesions) and physical examination describing lesion appearance and distribution. RAS is usually a clinical diagnosis. Biopsy is typically not required, but it may be helpful in severe, persistent, or atypical cases to exclude other mucosal pathology or malignancy. If RAS is severe, persistent, or begins later in life, laboratory evaluation is reasonable to look for underlying contributors. Suggested labs include CBC, inflammatory markers (ESR, CRP), and nutritional studies such as vitamin B12, folate, and iron. Imaging studies are not indicated.
Treatment is primarily supportive and stepwise, because etiology is unclear and there is no curative medication. Good oral hygiene and avoidance of triggers (trauma, braces/hardware irritation, known food/drug triggers) are foundational. For symptomatic relief, topical anesthetics can be used, especially before meals, including 2% viscous lidocaine applied directly or “swish and spit,” and diphenhydramine liquid (12.5 mg/5 mL) using 5 mL swish and spit. Topical corticosteroids are first-line for mild to moderate RAS, such as dexamethasone elixir (0.5 mg/5 mL) with 5 mL swish and spit three to four times daily. In more severe cases or in immunosuppressed patients, topical antimicrobials may reduce secondary infection risk, including chlorhexidine 0.12% mouth rinse (15 mL swish and spit twice daily) and nystatin suspension (“swish and swallow” four times daily) when candidal overgrowth is a concern.
For patients who do not improve with topical therapy alone or who have severe disease, oral options may be used. Oral prednisone 20–40 mg daily for 4–7 days is the first-line treatment for acute severe RAS. Montelukast 10 mg daily may be considered as a safer alternative in severe RAS that is not well controlled with oral corticosteroids. Complementary options sometimes used as adjuncts include ascorbic acid 2000 mg/day (particularly for minor RAS) and zinc 50 mg/day to support wound healing. Nonpharmacologic adjuncts like laser therapy or chemical cauterization have been used to reduce acute pain, but overall efficacy is unclear.
Disposition is generally reassuring because aphthous ulcers and RAS are typically self-resolving, with average healing around 2 weeks, and prognosis is excellent. However, clinicians should consider alternative causes when ulcers are persistent, unusually severe, occur outside typical patterns, or are accompanied by systemic symptoms. Referral is recommended when another diagnosis is suspected; severe or persistent RAS may be referred to dermatology. Prevention focuses on good oral hygiene, trigger avoidance, and ensuring adequate nutrition, especially addressing deficiencies. Patient and family education should emphasize pain control (using topical anesthetics before eating/drinking to prevent dehydration and before oral hygiene), avoidance of irritating foods such as citrus, spicy, salty foods, and return precautions including worsening pain, spreading ulcers beyond oral mucocutaneous membranes, fever, decreased oral intake, or other systemic symptoms.
Aphthous ulcers are small, painful ulcers that occur predominantly on the oral mucosa. They are also known as aphthae, canker sores, recurrent oral aphthae, or recurrent aphthous stomatitis (RAS). The ICD-10CM code is K12.0 (Recurrent oral aphthae). The incidence is unknown, but prevalence is common—about one in five people experience them at some point in life. They most commonly affect women, adolescents, and adults under 40, with peak incidence in adolescence. Risk factors include genetic predisposition, stress, local trauma, tobacco exposure, viral or bacterial infections, nutritional deficiencies, and endocrine or autoimmune diseases. Genetics play a role, as up to 40% of patients report a family history.
Clinically, aphthous ulcers are typically small round or oval lesions with a gray-white or yellow pseudomembranous base and an erythematous (red) halo. They can occur at any age, often beginning in childhood and commonly improving by the third decade. The exact cause is not well understood and is likely multifactorial, with contributors such as trauma (biting, dental appliances/braces), nutritional deficiencies, infections, medications, food hypersensitivity, hormonal/endocrine changes, and tobacco use. When ulcers recur without an identified systemic disease, the condition is termed recurrent aphthous stomatitis (RAS).
RAS is divided into three subtypes. Minor RAS (about 80%) presents as 1 to 5 shallow ulcers that are <10 mm, usually on the lips, buccal mucosa, tongue, and floor of mouth, they typically resolve in 4 to 14 days without scarring. major ras (about 10%) features larger (>10 mm), deeper ulcers that may be more numerous and extend to areas like the gingiva and pharyngeal mucosa; they last weeks to months and have a higher risk of scarring. Herpetiform RAS (about 10%) consists of many very painful small ulcers (1–3 mm) occurring in large numbers (5 to 100), often involving the floor of mouth and ventral tongue; ulcers may cluster and coalesce into larger irregular lesions, generally resolving within 14 days and usually without visible scarring. Despite the name, herpetiform RAS is not caused by herpes.
The differential diagnosis is broad, especially when ulcers are severe, persistent, atypical, or associated with systemic findings. Important considerations include Behçet syndrome, MAGIC syndrome, systemic lupus erythematosus, gluten-sensitive enteropathy, inflammatory bowel disease, HIV infection, herpes simplex virus, cyclic neutropenia, PFAPA syndrome, hyperimmunoglobulin D syndrome, agranulocytosis, autoimmune bullous disease, oral erosive lichen planus, drug-induced mucosal ulcers, contact dermatitis, and squamous cell carcinoma. Differentiating HSV from aphthous ulcers is useful: HSV tends to involve keratinized tissue, often has a vesicle phase and prodrome, whereas aphthous ulcers usually occur on movable, nonkeratinized mucosa, lack vesicles, and have nonspecific biopsy findings if biopsied.
Workup begins with a detailed clinical history (associated symptoms, family history, frequency, number/size, duration, location, and presence of any nonoral lesions) and physical examination describing lesion appearance and distribution. RAS is usually a clinical diagnosis. Biopsy is typically not required, but it may be helpful in severe, persistent, or atypical cases to exclude other mucosal pathology or malignancy. If RAS is severe, persistent, or begins later in life, laboratory evaluation is reasonable to look for underlying contributors. Suggested labs include CBC, inflammatory markers (ESR, CRP), and nutritional studies such as vitamin B12, folate, and iron. Imaging studies are not indicated.
Treatment is primarily supportive and stepwise, because etiology is unclear and there is no curative medication. Good oral hygiene and avoidance of triggers (trauma, braces/hardware irritation, known food/drug triggers) are foundational. For symptomatic relief, topical anesthetics can be used, especially before meals, including 2% viscous lidocaine applied directly or “swish and spit,” and diphenhydramine liquid (12.5 mg/5 mL) using 5 mL swish and spit. Topical corticosteroids are first-line for mild to moderate RAS, such as dexamethasone elixir (0.5 mg/5 mL) with 5 mL swish and spit three to four times daily. In more severe cases or in immunosuppressed patients, topical antimicrobials may reduce secondary infection risk, including chlorhexidine 0.12% mouth rinse (15 mL swish and spit twice daily) and nystatin suspension (“swish and swallow” four times daily) when candidal overgrowth is a concern.
For patients who do not improve with topical therapy alone or who have severe disease, oral options may be used. Oral prednisone 20–40 mg daily for 4–7 days is the first-line treatment for acute severe RAS. Montelukast 10 mg daily may be considered as a safer alternative in severe RAS that is not well controlled with oral corticosteroids. Complementary options sometimes used as adjuncts include ascorbic acid 2000 mg/day (particularly for minor RAS) and zinc 50 mg/day to support wound healing. Nonpharmacologic adjuncts like laser therapy or chemical cauterization have been used to reduce acute pain, but overall efficacy is unclear.
Disposition is generally reassuring because aphthous ulcers and RAS are typically self-resolving, with average healing around 2 weeks, and prognosis is excellent. However, clinicians should consider alternative causes when ulcers are persistent, unusually severe, occur outside typical patterns, or are accompanied by systemic symptoms. Referral is recommended when another diagnosis is suspected; severe or persistent RAS may be referred to dermatology. Prevention focuses on good oral hygiene, trigger avoidance, and ensuring adequate nutrition, especially addressing deficiencies. Patient and family education should emphasize pain control (using topical anesthetics before eating/drinking to prevent dehydration and before oral hygiene), avoidance of irritating foods such as citrus, spicy, salty foods, and return precautions including worsening pain, spreading ulcers beyond oral mucocutaneous membranes, fever, decreased oral intake, or other systemic symptoms.

