- Published on
Emergency And Acute Medicine – Inborn Errors Of Metabolism
Basics
Description Inborn errors of metabolism are inherited defects in metabolic pathways that lead to abnormal accumulation or deficiency of metabolites. More than 400 disorders are described, most presenting in infancy or childhood with variable and often nonspecific findings. Incidence ranges from 1:10,000 to 1:200,000 births. Major categories include amino acid disorders, urea cycle defects, organic acidemias, fatty acid oxidation defects, mitochondrial disorders, carbohydrate disorders, lysosomal storage diseases, peroxisomal disorders, and disorders of protein glycosylation. Pathophysiology relates directly to enzymatic or transport defects within metabolic pathways.
Etiology Genetic deficiency of enzymes or membrane transport systems involved in intermediary metabolism.
Diagnosis
Signs and symptoms Presentation may be acute and catastrophic or chronic and indolent. Neonates may initially be asymptomatic or present with hypothermia (mitochondrial defects), hypotonia or hypertonia (peroxisomal disorders), apnea (urea cycle defects, organic acidemias), seizures, vomiting, poor feeding, jaundice, hypoglycemia, dysmorphic features, or coma. Older untreated children may present with failure to thrive, dehydration, recurrent vomiting or diarrhea, food intolerance, lethargy, ataxia, seizures, or developmental delay.
History Review newborn screening results, dietary history, family history, consanguinity, and intercurrent illness.
Physical exam May reveal abnormal odor, altered mental status, tachypnea, dysmorphic facies, cataracts, cardiomyopathy, hepatosplenomegaly, dermatitis, or jaundice.
Essential workup Consider in any child with unexplained neurologic deterioration, persistent vomiting, metabolic acidosis, hypoglycemia, shock unresponsive to resuscitation, or failure to thrive.
Diagnosis tests and interpretation
Lab Bedside glucose, electrolytes, BUN/creatinine, CBC, calcium, liver function tests, bilirubin, coagulation studies, blood gas, lactate, pyruvate, uric acid, urinalysis, ammonia, serum amino acids, urine organic acids, and cultures as indicated.
Imaging CT head for altered mental status; chest radiograph as indicated.
Procedures Lumbar puncture when infection is a concern.
Differential diagnosis Sepsis, meningitis, encephalitis, dehydration, toxic ingestion, nonaccidental trauma, Reye syndrome, hepatic encephalopathy, endocrine disorders, renal failure, renal tubular acidosis, CNS mass lesions.
Treatment
Prehospital Prioritize ABCs and bedside glucose. Start IV glucose immediately; do not delay for fluids unless in shock. Avoid lactated Ringer solution. Keep patient NPO.
Initial stabilization Administer glucose (after Accu-Chek and thiamine when appropriate) and naloxone for altered mental status.
Emergency department management Secure airway, breathing, and circulation. Use normal saline for fluid boluses and avoid hypotonic fluids and lactated Ringer. Initiate IV glucose at 8–10 mg/kg/min (D10 at 1.5× maintenance) to prevent catabolism. Treat severe hypoglycemia with D25 bolus. Correct dehydration and acid–base abnormalities; consider bicarbonate only if pH <7.0. initiate dialysis for refractory severe acidosis or hyperammonemia. stop all oral intake initially. treat hyperammonemia urgently with ammonia-scavenging agents (arginine, sodium benzoate, phenylacetate, phenylbutyrate) in consultation a metabolic specialist. identify and precipitating infections.< />pan>
Medication D25 2–4 mL/kg IV for hypoglycemia, sodium bicarbonate 1–2 mEq/kg IV for severe acidosis, disease-specific therapies such as levocarnitine or pyridoxine as indicated.
First line IV glucose infusion with normal saline as needed for shock.
Second line Bicarbonate therapy for pH <7.0 and hemodialysis when indicated.< />pan>
Follow-up and disposition
Admission criteria All infants and children with suspected inborn errors of metabolism, significant ketosis, inability to tolerate oral intake, altered mental status, severe or persistent acidosis, refractory hypoglycemia, or hyperammonemia; ICU care for severe presentations. Transfer to a specialized pediatric center is often required.
Discharge criteria Normal mental status, adequate hydration, reassuring laboratory results, no significant intercurrent illness, and close follow-up arranged.
Follow-up recommendations Ongoing care with a metabolic disease specialist and primary care physician.
Pearls and pitfalls Always consider metabolic disease in unexplained pediatric decompensation. Do not delay glucose infusion. Avoid lactated Ringer solution. Use bicarbonate cautiously and only for severe acidosis. Early dialysis can be lifesaving in hyperammonemia.
Basics
Description Inborn errors of metabolism are inherited defects in metabolic pathways that lead to abnormal accumulation or deficiency of metabolites. More than 400 disorders are described, most presenting in infancy or childhood with variable and often nonspecific findings. Incidence ranges from 1:10,000 to 1:200,000 births. Major categories include amino acid disorders, urea cycle defects, organic acidemias, fatty acid oxidation defects, mitochondrial disorders, carbohydrate disorders, lysosomal storage diseases, peroxisomal disorders, and disorders of protein glycosylation. Pathophysiology relates directly to enzymatic or transport defects within metabolic pathways.
Etiology Genetic deficiency of enzymes or membrane transport systems involved in intermediary metabolism.
Diagnosis
Signs and symptoms Presentation may be acute and catastrophic or chronic and indolent. Neonates may initially be asymptomatic or present with hypothermia (mitochondrial defects), hypotonia or hypertonia (peroxisomal disorders), apnea (urea cycle defects, organic acidemias), seizures, vomiting, poor feeding, jaundice, hypoglycemia, dysmorphic features, or coma. Older untreated children may present with failure to thrive, dehydration, recurrent vomiting or diarrhea, food intolerance, lethargy, ataxia, seizures, or developmental delay.
History Review newborn screening results, dietary history, family history, consanguinity, and intercurrent illness.
Physical exam May reveal abnormal odor, altered mental status, tachypnea, dysmorphic facies, cataracts, cardiomyopathy, hepatosplenomegaly, dermatitis, or jaundice.
Essential workup Consider in any child with unexplained neurologic deterioration, persistent vomiting, metabolic acidosis, hypoglycemia, shock unresponsive to resuscitation, or failure to thrive.
Diagnosis tests and interpretation
Lab Bedside glucose, electrolytes, BUN/creatinine, CBC, calcium, liver function tests, bilirubin, coagulation studies, blood gas, lactate, pyruvate, uric acid, urinalysis, ammonia, serum amino acids, urine organic acids, and cultures as indicated.
Imaging CT head for altered mental status; chest radiograph as indicated.
Procedures Lumbar puncture when infection is a concern.
Differential diagnosis Sepsis, meningitis, encephalitis, dehydration, toxic ingestion, nonaccidental trauma, Reye syndrome, hepatic encephalopathy, endocrine disorders, renal failure, renal tubular acidosis, CNS mass lesions.
Treatment
Prehospital Prioritize ABCs and bedside glucose. Start IV glucose immediately; do not delay for fluids unless in shock. Avoid lactated Ringer solution. Keep patient NPO.
Initial stabilization Administer glucose (after Accu-Chek and thiamine when appropriate) and naloxone for altered mental status.
Emergency department management Secure airway, breathing, and circulation. Use normal saline for fluid boluses and avoid hypotonic fluids and lactated Ringer. Initiate IV glucose at 8–10 mg/kg/min (D10 at 1.5× maintenance) to prevent catabolism. Treat severe hypoglycemia with D25 bolus. Correct dehydration and acid–base abnormalities; consider bicarbonate only if pH <7.0. initiate dialysis for refractory severe acidosis or hyperammonemia. stop all oral intake initially. treat hyperammonemia urgently with ammonia-scavenging agents (arginine, sodium benzoate, phenylacetate, phenylbutyrate) in consultation a metabolic specialist. identify and precipitating infections.< />pan>
Medication D25 2–4 mL/kg IV for hypoglycemia, sodium bicarbonate 1–2 mEq/kg IV for severe acidosis, disease-specific therapies such as levocarnitine or pyridoxine as indicated.
First line IV glucose infusion with normal saline as needed for shock.
Second line Bicarbonate therapy for pH <7.0 and hemodialysis when indicated.< />pan>
Follow-up and disposition
Admission criteria All infants and children with suspected inborn errors of metabolism, significant ketosis, inability to tolerate oral intake, altered mental status, severe or persistent acidosis, refractory hypoglycemia, or hyperammonemia; ICU care for severe presentations. Transfer to a specialized pediatric center is often required.
Discharge criteria Normal mental status, adequate hydration, reassuring laboratory results, no significant intercurrent illness, and close follow-up arranged.
Follow-up recommendations Ongoing care with a metabolic disease specialist and primary care physician.
Pearls and pitfalls Always consider metabolic disease in unexplained pediatric decompensation. Do not delay glucose infusion. Avoid lactated Ringer solution. Use bicarbonate cautiously and only for severe acidosis. Early dialysis can be lifesaving in hyperammonemia.

- Published on
Emergency And Acute Medicine – Impetigo
Basics
Description Impetigo is a common superficial bacterial skin infection. It may be a primary infection of minor skin breaks or a secondary infection of preexisting dermatoses (impetiginization). It is most prevalent in children aged 2–5 years and occurs more often in warm, humid climates and summer months. Predisposing factors include minor trauma (especially around the nose), insect bites, burns, diabetes mellitus, HIV infection, varicella, and chronic skin disease. Complications include acute poststreptococcal glomerulonephritis (1–5% of nonbullous cases), cellulitis, sepsis, endocarditis, toxic shock syndrome, and staphylococcal scalded skin syndrome.
Etiology
Classic (nonbullous) impetigo results from bacterial entry through disrupted skin and is caused by Staphylococcus aureus, group A β-hemolytic streptococci, or both. Bullous impetigo is caused exclusively by S. aureus producing exfoliative toxins (A, B, D) that cleave desmoglein 1, leading to intraepidermal blistering.
Diagnosis
Signs and symptoms Nonbullous impetigo begins as erythematous macules or papules that evolve into vesicles or pustules, which rupture and form characteristic honey-colored crusts. Lesions are highly contagious and often pruritic. Regional lymphadenopathy may occur; systemic symptoms are uncommon. Bullous impetigo presents with flaccid bullae containing clear or yellow fluid that rupture easily, leaving erythematous bases; Nikolsky sign is absent. Fever and systemic symptoms are rare.
Physical exam Lesions most commonly involve the face, extremities, and scalp. Diagnosis is clinical based on appearance and distribution.
Essential workup
Routine laboratory testing is not required. Consider bacterial culture of bullae or pustules for refractory disease, outbreak investigation, or suspected methicillin-resistant S. aureus.
Diagnosis tests and interpretation
Lab Anti–DNase B titers may be elevated in poststreptococcal glomerulonephritis. Urinalysis is indicated if hematuria or edema suggests nephritis.
Imaging and procedures Not routinely indicated; biopsy is rarely necessary.
Differential diagnosis
Herpes simplex, varicella zoster, atopic or contact dermatitis, dermatophytosis, candidiasis, scabies, folliculitis, erysipelas, bullous pemphigoid, pemphigus vulgaris, staphylococcal scalded skin syndrome, Stevens–Johnson syndrome, thermal burns, toxic epidermal necrolysis, and cutaneous anthrax.
Treatment
Prehospital Cover lesions and use universal precautions to prevent transmission.
Initial stabilization Impetigo is rarely life-threatening in otherwise healthy patients.
Emergency department management Local care includes gentle cleansing, removal of crusts, and wet dressings. Small, localized nonbullous lesions may be treated with topical antibiotics alone. Extensive disease, bullous impetigo, lymphadenopathy, systemic symptoms, or outbreak settings require systemic antibiotics. Empiric therapy should cover S. aureus and streptococci; adjust for MRSA based on local resistance patterns.
Medication
Topical (nonbullous only): mupirocin 2% TID for 10 days or retapamulin 1% BID for 5 days.
Oral options (10 days unless noted): amoxicillin–clavulanate, cephalexin, dicloxacillin, clindamycin, azithromycin (5-day course), or trimethoprim–sulfamethoxazole when MRSA suspected. Doxycycline may be used for MRSA in patients ≥8 years.
Follow-up and disposition
Admission criteria Rare; consider for widespread disease, extensive bullae or denuded skin, dehydration, treatment failure, toxic appearance, immunocompromise, or neonates.
Discharge criteria Nontoxic patients with reliable caregivers and ability to comply with therapy.
Follow-up recommendations Primary care follow-up to ensure resolution. Return for lack of improvement or signs of nephritis (hematuria, periorbital or leg edema).
Pearls and pitfalls
Use systemic antibiotics for bullous disease or lymphadenopathy. Rising antibiotic resistance limits older regimens; suspect mupirocin resistance with treatment failure and switch agents. Treat affected household contacts simultaneously to prevent reinfection.
Basics
Description Impetigo is a common superficial bacterial skin infection. It may be a primary infection of minor skin breaks or a secondary infection of preexisting dermatoses (impetiginization). It is most prevalent in children aged 2–5 years and occurs more often in warm, humid climates and summer months. Predisposing factors include minor trauma (especially around the nose), insect bites, burns, diabetes mellitus, HIV infection, varicella, and chronic skin disease. Complications include acute poststreptococcal glomerulonephritis (1–5% of nonbullous cases), cellulitis, sepsis, endocarditis, toxic shock syndrome, and staphylococcal scalded skin syndrome.
Etiology
Classic (nonbullous) impetigo results from bacterial entry through disrupted skin and is caused by Staphylococcus aureus, group A β-hemolytic streptococci, or both. Bullous impetigo is caused exclusively by S. aureus producing exfoliative toxins (A, B, D) that cleave desmoglein 1, leading to intraepidermal blistering.
Diagnosis
Signs and symptoms Nonbullous impetigo begins as erythematous macules or papules that evolve into vesicles or pustules, which rupture and form characteristic honey-colored crusts. Lesions are highly contagious and often pruritic. Regional lymphadenopathy may occur; systemic symptoms are uncommon. Bullous impetigo presents with flaccid bullae containing clear or yellow fluid that rupture easily, leaving erythematous bases; Nikolsky sign is absent. Fever and systemic symptoms are rare.
Physical exam Lesions most commonly involve the face, extremities, and scalp. Diagnosis is clinical based on appearance and distribution.
Essential workup
Routine laboratory testing is not required. Consider bacterial culture of bullae or pustules for refractory disease, outbreak investigation, or suspected methicillin-resistant S. aureus.
Diagnosis tests and interpretation
Lab Anti–DNase B titers may be elevated in poststreptococcal glomerulonephritis. Urinalysis is indicated if hematuria or edema suggests nephritis.
Imaging and procedures Not routinely indicated; biopsy is rarely necessary.
Differential diagnosis
Herpes simplex, varicella zoster, atopic or contact dermatitis, dermatophytosis, candidiasis, scabies, folliculitis, erysipelas, bullous pemphigoid, pemphigus vulgaris, staphylococcal scalded skin syndrome, Stevens–Johnson syndrome, thermal burns, toxic epidermal necrolysis, and cutaneous anthrax.
Treatment
Prehospital Cover lesions and use universal precautions to prevent transmission.
Initial stabilization Impetigo is rarely life-threatening in otherwise healthy patients.
Emergency department management Local care includes gentle cleansing, removal of crusts, and wet dressings. Small, localized nonbullous lesions may be treated with topical antibiotics alone. Extensive disease, bullous impetigo, lymphadenopathy, systemic symptoms, or outbreak settings require systemic antibiotics. Empiric therapy should cover S. aureus and streptococci; adjust for MRSA based on local resistance patterns.
Medication
Topical (nonbullous only): mupirocin 2% TID for 10 days or retapamulin 1% BID for 5 days.
Oral options (10 days unless noted): amoxicillin–clavulanate, cephalexin, dicloxacillin, clindamycin, azithromycin (5-day course), or trimethoprim–sulfamethoxazole when MRSA suspected. Doxycycline may be used for MRSA in patients ≥8 years.
Follow-up and disposition
Admission criteria Rare; consider for widespread disease, extensive bullae or denuded skin, dehydration, treatment failure, toxic appearance, immunocompromise, or neonates.
Discharge criteria Nontoxic patients with reliable caregivers and ability to comply with therapy.
Follow-up recommendations Primary care follow-up to ensure resolution. Return for lack of improvement or signs of nephritis (hematuria, periorbital or leg edema).
Pearls and pitfalls
Use systemic antibiotics for bullous disease or lymphadenopathy. Rising antibiotic resistance limits older regimens; suspect mupirocin resistance with treatment failure and switch agents. Treat affected household contacts simultaneously to prevent reinfection.

- Published on
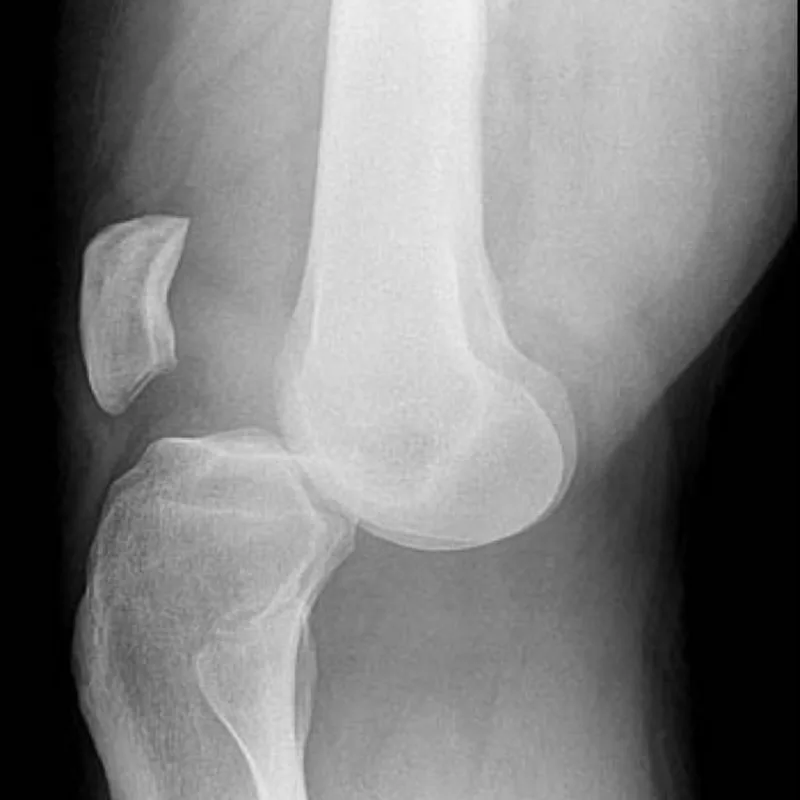
Emergency And Acute Medicine – Knee Dislocation
Basics Description
Knee dislocation is defined by displacement of the tibia relative to the distal femur and represents a true orthopedic emergency because of the high risk of vascular and neurologic injury. Anterior dislocation is the most common type, accounting for approximately 60% of cases, and typically results from hyperextension. Rupture of the posterior capsule may occur at 30° of hyperextension, with disruption of the posterior cruciate ligament and potential popliteal artery injury at greater degrees. Posterior dislocation usually results from a direct blow to the anterior tibia with the knee flexed, such as a dashboard injury. Medial and lateral dislocations occur from varus or valgus stress, respectively, and are associated with multiligamentous injuries.
Popliteal artery injury occurs in up to 35% of knee dislocations and is the most limb-threatening complication. Anterior dislocations commonly cause traction-related intimal injury with delayed thrombosis, whereas posterior dislocations are more likely to cause direct arterial transection and immediate thrombosis. Peroneal nerve injury is less common but clinically significant and often accompanies arterial injury, particularly in medial or rotational dislocations.
Etiology
Knee dislocations typically result from high-energy mechanisms such as motor vehicle collisions, auto–pedestrian trauma, or athletic injuries, most commonly football.
Diagnosis Signs And Symptoms
Patients may present with a grossly deformed or unstable knee, though spontaneous reduction can mask deformity. Absence of distal pulses or signs of ischemia such as pallor, paresthesia, pain, paralysis, or poikilothermia raise immediate concern for popliteal artery injury. Neurologic deficits may include decreased sensation in the first web space or inability to dorsiflex the foot. A careful history of the mechanism of injury and repeated neurovascular examinations are essential.
Essential Workup
Evaluation requires a detailed history and thorough physical examination with emphasis on vascular and neurologic status. Distal pulses should be assessed by palpation and Doppler, capillary refill documented, and ankle–brachial index calculated. Neurologic examination should assess peroneal nerve function. Repeat examinations are mandatory, especially after reduction.
Diagnosis Tests And Interpretation
AP and lateral knee radiographs are required to confirm alignment and exclude associated fractures. MRI within one week is useful for defining ligamentous injury. An ABI ≥0.9 suggests low likelihood of major arterial injury, but abnormal findings warrant further evaluation. Vascular ultrasound or arteriography should be considered when pulses are diminished, ischemic symptoms persist, or peroneal nerve injury is present.
Differential Diagnosis
Conditions to consider include tibial plateau fracture, supracondylar femoral fracture, and ligamentous or tendinous avulsion injuries.
Treatment Pre Hospital
Initial management focuses on airway, breathing, and circulation. Distal pulses and motor function must be documented. The knee should be splinted in slight flexion to minimize popliteal artery tension.
Initial Stabilization Therapy
Address ABCs, particularly in polytrauma patients. Hypotension should be corrected, as it may obscure vascular compromise. Immediate closed reduction is indicated for any evidence of limb ischemia, with early orthopedic and vascular consultation.
Ed Treatment Procedures
Closed reduction is performed using longitudinal traction with gentle realignment, avoiding pressure in the popliteal fossa. The knee is immobilized in 15–20° of flexion. Neurovascular status must be reassessed immediately after reduction and monitored frequently. IV analgesia should be provided, and surgical consultation obtained for open injuries, vascular compromise, or irreducible dislocations.
Medication
IV narcotic analgesia is first-line. Oral medications should be avoided due to the high likelihood of operative intervention.
Follow-Up Disposition
All patients with knee dislocation require admission for observation and management of potential vascular injury. Discharge from the ED is not appropriate.
Follow-Up Recommendations
Orthopedic follow-up is required for staged ligamentous repair, typically delayed until swelling subsides. Vascular surgery follow-up is necessary if popliteal artery injury is identified.
Pearls And Pitfalls
Failure to restore popliteal artery flow within 6–8 hours carries an amputation risk approaching 90%. Peroneal nerve injury has a poor prognosis for recovery. Delayed compartment syndrome may occur and requires vigilant monitoring.
Basics Description
Knee dislocation is defined by displacement of the tibia relative to the distal femur and represents a true orthopedic emergency because of the high risk of vascular and neurologic injury. Anterior dislocation is the most common type, accounting for approximately 60% of cases, and typically results from hyperextension. Rupture of the posterior capsule may occur at 30° of hyperextension, with disruption of the posterior cruciate ligament and potential popliteal artery injury at greater degrees. Posterior dislocation usually results from a direct blow to the anterior tibia with the knee flexed, such as a dashboard injury. Medial and lateral dislocations occur from varus or valgus stress, respectively, and are associated with multiligamentous injuries.
Popliteal artery injury occurs in up to 35% of knee dislocations and is the most limb-threatening complication. Anterior dislocations commonly cause traction-related intimal injury with delayed thrombosis, whereas posterior dislocations are more likely to cause direct arterial transection and immediate thrombosis. Peroneal nerve injury is less common but clinically significant and often accompanies arterial injury, particularly in medial or rotational dislocations.
Etiology
Knee dislocations typically result from high-energy mechanisms such as motor vehicle collisions, auto–pedestrian trauma, or athletic injuries, most commonly football.
Diagnosis Signs And Symptoms
Patients may present with a grossly deformed or unstable knee, though spontaneous reduction can mask deformity. Absence of distal pulses or signs of ischemia such as pallor, paresthesia, pain, paralysis, or poikilothermia raise immediate concern for popliteal artery injury. Neurologic deficits may include decreased sensation in the first web space or inability to dorsiflex the foot. A careful history of the mechanism of injury and repeated neurovascular examinations are essential.
Essential Workup
Evaluation requires a detailed history and thorough physical examination with emphasis on vascular and neurologic status. Distal pulses should be assessed by palpation and Doppler, capillary refill documented, and ankle–brachial index calculated. Neurologic examination should assess peroneal nerve function. Repeat examinations are mandatory, especially after reduction.
Diagnosis Tests And Interpretation
AP and lateral knee radiographs are required to confirm alignment and exclude associated fractures. MRI within one week is useful for defining ligamentous injury. An ABI ≥0.9 suggests low likelihood of major arterial injury, but abnormal findings warrant further evaluation. Vascular ultrasound or arteriography should be considered when pulses are diminished, ischemic symptoms persist, or peroneal nerve injury is present.
Differential Diagnosis
Conditions to consider include tibial plateau fracture, supracondylar femoral fracture, and ligamentous or tendinous avulsion injuries.
Treatment Pre Hospital
Initial management focuses on airway, breathing, and circulation. Distal pulses and motor function must be documented. The knee should be splinted in slight flexion to minimize popliteal artery tension.
Initial Stabilization Therapy
Address ABCs, particularly in polytrauma patients. Hypotension should be corrected, as it may obscure vascular compromise. Immediate closed reduction is indicated for any evidence of limb ischemia, with early orthopedic and vascular consultation.
Ed Treatment Procedures
Closed reduction is performed using longitudinal traction with gentle realignment, avoiding pressure in the popliteal fossa. The knee is immobilized in 15–20° of flexion. Neurovascular status must be reassessed immediately after reduction and monitored frequently. IV analgesia should be provided, and surgical consultation obtained for open injuries, vascular compromise, or irreducible dislocations.
Medication
IV narcotic analgesia is first-line. Oral medications should be avoided due to the high likelihood of operative intervention.
Follow-Up Disposition
All patients with knee dislocation require admission for observation and management of potential vascular injury. Discharge from the ED is not appropriate.
Follow-Up Recommendations
Orthopedic follow-up is required for staged ligamentous repair, typically delayed until swelling subsides. Vascular surgery follow-up is necessary if popliteal artery injury is identified.
Pearls And Pitfalls
Failure to restore popliteal artery flow within 6–8 hours carries an amputation risk approaching 90%. Peroneal nerve injury has a poor prognosis for recovery. Delayed compartment syndrome may occur and requires vigilant monitoring.
- Published on
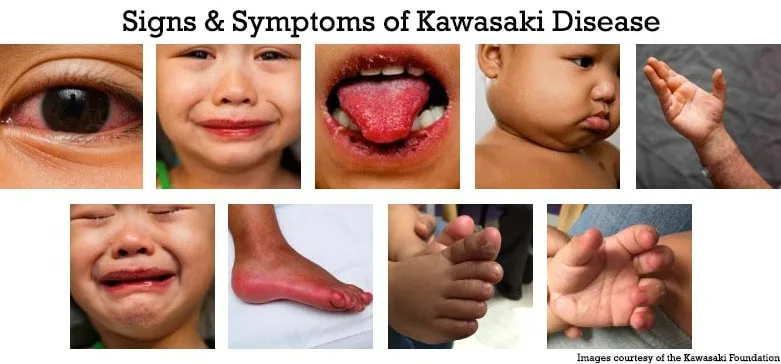
Emergency and Acute Medicine – Kawasaki disease
Kawasaki disease is an acute, self-limited inflammatory illness involving multiple organ systems and is the leading cause of acquired heart disease in children in developed countries. It is a medium-vessel vasculitis with particular predilection for the coronary arteries. Acute cardiac complications include myocarditis, pericarditis, and coronary artery aneurysms, which may later progress to stenosis or thrombosis; giant aneurysms carry a risk of rupture. The disease progresses through three stages: an acute phase lasting 1–2 weeks characterized by fever and systemic inflammation; a subacute phase extending to approximately 4 weeks marked by desquamation, thrombocytosis, and the highest risk of sudden death from coronary involvement; and a convalescent phase lasting 6–8 weeks during which symptoms resolve and inflammatory markers normalize.
Epidemiologically, 80% of cases occur in children younger than 4 years, with a peak incidence between 1 and 2 years of age. The disease is rare in infants younger than 3 months, though adult cases have been reported. Males are affected more often than females, and children of Asian descent have the highest incidence, suggesting a genetic predisposition. Delayed diagnosis, prolonged fever, male sex, extremes of age, elevated inflammatory markers, anemia, hypoalbuminemia, and abnormal initial echocardiography increase the risk of coronary artery aneurysm formation. Approximately 10–15% of patients are resistant to standard therapy.
The etiology remains unknown, but the disease is thought to represent an abnormal immune response to an infectious trigger in genetically susceptible individuals. Seasonal clustering and epidemic patterns support this hypothesis, with increased incidence in winter and early spring.
Diagnosis is clinical. Classic Kawasaki disease is defined by fever lasting at least 5 days in combination with four of five features: bilateral nonexudative conjunctival injection, oral mucosal changes (such as strawberry tongue or cracked lips), polymorphous rash, extremity changes (erythema, edema, or desquamation), and cervical lymphadenopathy greater than 1.5 cm. Incomplete or atypical Kawasaki disease should be suspected in children with prolonged fever and fewer criteria accompanied by elevated ESR or CRP and supportive laboratory findings such as anemia, hypoalbuminemia, elevated ALT, leukocytosis, thrombocytosis after day 7, or sterile pyuria.
Clinical presentation typically includes abrupt onset of high, spiking fever persisting longer than 5 days. Children often appear irritable and ill. Ocular findings include conjunctivitis without exudate and photophobia. Oral findings include erythema, fissured lips, and strawberry tongue. Skin findings are variable and usually involve the trunk, while extremity changes include painful edema and erythema followed later by desquamation of the fingers and toes. Gastrointestinal symptoms such as vomiting, diarrhea, abdominal pain, and gallbladder hydrops may occur. Cardiac manifestations include myocarditis, pericarditis, heart failure, and, later, coronary artery aneurysms.
Evaluation requires a high index of suspicion in any febrile child with rash. Laboratory findings typically include elevated ESR and CRP, leukocytosis with left shift, normocytic anemia, thrombocytosis in the subacute phase, sterile pyuria, and mild transaminase elevation. Blood, urine, CSF, and throat cultures are negative. Echocardiography is essential to assess coronary artery involvement and should be performed at diagnosis, at 2–3 weeks, and again at 6–8 weeks. ECG may be indicated if myocardial involvement is suspected.
The differential diagnosis includes viral illnesses such as adenovirus, measles, Epstein–Barr virus, and influenza; bacterial infections such as scarlet fever, staphylococcal scalded skin syndrome, and rickettsial disease; immune-mediated conditions including Stevens–Johnson syndrome and serum sickness; and other vasculitides or connective tissue diseases.
Management focuses on early treatment to prevent cardiac complications. All patients meeting diagnostic criteria should be admitted. Initial therapy consists of intravenous immunoglobulin and high-dose aspirin, ideally within the first 10 days of illness, which reduces the incidence of coronary artery aneurysms from approximately 20–25% to 2–4%. Cardiology consultation is required, and myocardial infarction is treated similarly to adults if it occurs.
First-line treatment includes IVIG at 2 g/kg administered over 10–12 hours, with repeat dosing for persistent or recrudescent fever. High-dose aspirin is given during the acute phase for anti-inflammatory effect, followed by low-dose aspirin for antiplatelet therapy during the convalescent period. Patients who fail two doses of IVIG may require corticosteroids or other immunomodulatory therapies such as infliximab or cyclosporine.
All patients diagnosed with Kawasaki disease require hospitalization, while nontoxic children who do not meet diagnostic criteria may be discharged with close follow-up. Long-term cardiology follow-up is mandatory for monitoring coronary artery involvement.
Early recognition and treatment are critical, as prompt IVIG and aspirin therapy prevent coronary aneurysm formation in the vast majority of patients. Kawasaki disease should be strongly considered in children with persistent fever and repeated emergency visits, and incomplete presentations must not be overlooked.
Kawasaki disease is an acute, self-limited inflammatory illness involving multiple organ systems and is the leading cause of acquired heart disease in children in developed countries. It is a medium-vessel vasculitis with particular predilection for the coronary arteries. Acute cardiac complications include myocarditis, pericarditis, and coronary artery aneurysms, which may later progress to stenosis or thrombosis; giant aneurysms carry a risk of rupture. The disease progresses through three stages: an acute phase lasting 1–2 weeks characterized by fever and systemic inflammation; a subacute phase extending to approximately 4 weeks marked by desquamation, thrombocytosis, and the highest risk of sudden death from coronary involvement; and a convalescent phase lasting 6–8 weeks during which symptoms resolve and inflammatory markers normalize.
Epidemiologically, 80% of cases occur in children younger than 4 years, with a peak incidence between 1 and 2 years of age. The disease is rare in infants younger than 3 months, though adult cases have been reported. Males are affected more often than females, and children of Asian descent have the highest incidence, suggesting a genetic predisposition. Delayed diagnosis, prolonged fever, male sex, extremes of age, elevated inflammatory markers, anemia, hypoalbuminemia, and abnormal initial echocardiography increase the risk of coronary artery aneurysm formation. Approximately 10–15% of patients are resistant to standard therapy.
The etiology remains unknown, but the disease is thought to represent an abnormal immune response to an infectious trigger in genetically susceptible individuals. Seasonal clustering and epidemic patterns support this hypothesis, with increased incidence in winter and early spring.
Diagnosis is clinical. Classic Kawasaki disease is defined by fever lasting at least 5 days in combination with four of five features: bilateral nonexudative conjunctival injection, oral mucosal changes (such as strawberry tongue or cracked lips), polymorphous rash, extremity changes (erythema, edema, or desquamation), and cervical lymphadenopathy greater than 1.5 cm. Incomplete or atypical Kawasaki disease should be suspected in children with prolonged fever and fewer criteria accompanied by elevated ESR or CRP and supportive laboratory findings such as anemia, hypoalbuminemia, elevated ALT, leukocytosis, thrombocytosis after day 7, or sterile pyuria.
Clinical presentation typically includes abrupt onset of high, spiking fever persisting longer than 5 days. Children often appear irritable and ill. Ocular findings include conjunctivitis without exudate and photophobia. Oral findings include erythema, fissured lips, and strawberry tongue. Skin findings are variable and usually involve the trunk, while extremity changes include painful edema and erythema followed later by desquamation of the fingers and toes. Gastrointestinal symptoms such as vomiting, diarrhea, abdominal pain, and gallbladder hydrops may occur. Cardiac manifestations include myocarditis, pericarditis, heart failure, and, later, coronary artery aneurysms.
Evaluation requires a high index of suspicion in any febrile child with rash. Laboratory findings typically include elevated ESR and CRP, leukocytosis with left shift, normocytic anemia, thrombocytosis in the subacute phase, sterile pyuria, and mild transaminase elevation. Blood, urine, CSF, and throat cultures are negative. Echocardiography is essential to assess coronary artery involvement and should be performed at diagnosis, at 2–3 weeks, and again at 6–8 weeks. ECG may be indicated if myocardial involvement is suspected.
The differential diagnosis includes viral illnesses such as adenovirus, measles, Epstein–Barr virus, and influenza; bacterial infections such as scarlet fever, staphylococcal scalded skin syndrome, and rickettsial disease; immune-mediated conditions including Stevens–Johnson syndrome and serum sickness; and other vasculitides or connective tissue diseases.
Management focuses on early treatment to prevent cardiac complications. All patients meeting diagnostic criteria should be admitted. Initial therapy consists of intravenous immunoglobulin and high-dose aspirin, ideally within the first 10 days of illness, which reduces the incidence of coronary artery aneurysms from approximately 20–25% to 2–4%. Cardiology consultation is required, and myocardial infarction is treated similarly to adults if it occurs.
First-line treatment includes IVIG at 2 g/kg administered over 10–12 hours, with repeat dosing for persistent or recrudescent fever. High-dose aspirin is given during the acute phase for anti-inflammatory effect, followed by low-dose aspirin for antiplatelet therapy during the convalescent period. Patients who fail two doses of IVIG may require corticosteroids or other immunomodulatory therapies such as infliximab or cyclosporine.
All patients diagnosed with Kawasaki disease require hospitalization, while nontoxic children who do not meet diagnostic criteria may be discharged with close follow-up. Long-term cardiology follow-up is mandatory for monitoring coronary artery involvement.
Early recognition and treatment are critical, as prompt IVIG and aspirin therapy prevent coronary aneurysm formation in the vast majority of patients. Kawasaki disease should be strongly considered in children with persistent fever and repeated emergency visits, and incomplete presentations must not be overlooked.
- Published on
Emergency and Acute Medicine – Isopropanol poisoning
Isopropanol is a central nervous system depressant that is two to three times more potent than ethanol. It is rapidly absorbed after ingestion and is metabolized by alcohol dehydrogenase to acetone, which is also a CNS depressant. Unlike other toxic alcohols, isopropanol is ketogenic but does not cause a significant metabolic acidosis. Acetone is eliminated by the lungs and kidneys. The half-life of isopropanol ranges from 3 to 16 hours, while acetone has a longer half-life of 7.5 to 26 hours. Concomitant ethanol ingestion prolongs the half-life of isopropanol but not acetone.
Isopropanol is a clear, colorless, volatile liquid with a faint acetone odor and bitter taste, commonly found as 70% rubbing alcohol. It is present in many household and industrial products including disinfectants, hand sanitizers, solvents, window cleaners, paint removers, detergents, antifreeze, and toiletries. The typical adult patient is a chronic alcoholic who ingests isopropanol after depleting ethanol supplies. Dermal, inhalational, and rectal exposures can also cause systemic toxicity. Accidental ingestions are common in young children.
Symptoms usually develop within 30 to 60 minutes of exposure. Neurologic manifestations include lethargy, inebriation, vertigo, ataxia, headache, weakness, respiratory depression, coma, and apnea, without the initial excitatory phase seen with ethanol. Gastrointestinal findings include nausea, vomiting, abdominal pain, gastritis, and hematemesis. Cardiovascular effects include hypotension, tachycardia, myocardial depression, and peripheral vasodilation. Pulmonary findings include respiratory depression and hemorrhagic tracheobronchitis. Skin and eye exposure may cause irritation or chemical burns.
Diagnosis is based on history of exposure and the characteristic odor of isopropanol or acetone on the breath. Laboratory evaluation typically shows ketosis without significant acidosis unless end-organ hypoperfusion is present. Hypoglycemia may occur, particularly in children. Acetone can falsely elevate serum creatinine, which normalizes as acetone is metabolized. Urinalysis shows ketones, and an elevated osmolar gap is common. Coma is associated with serum isopropanol levels greater than 150 mg/dL. Imaging is guided by clinical findings, with chest radiography used to assess for aspiration and head CT considered for possible trauma.
Management is primarily supportive, with attention to airway protection, breathing, and circulation. Hypotension is treated with intravenous fluids and vasopressors if needed. There is no specific antidote, and treatment with ethanol or fomepizole is contraindicated. Activated charcoal may be considered if coingestants are suspected. Skin and eye exposures require copious irrigation. Hemodialysis effectively removes isopropanol and acetone and is reserved for patients with refractory hypotension, severe toxicity, or serum levels greater than 400 mg/dL.
Patients with moderate to severe toxicity, including altered mental status or hypotension, require hospital admission. Asymptomatic or mildly intoxicated patients may be observed for 2 to 4 hours and discharged if symptoms resolve. Referral for alcohol detoxification or psychiatric evaluation is recommended for intentional ingestions.
Supportive care remains the cornerstone of treatment, and a key pitfall is inappropriate use of ethanol or fomepizole, which should be avoided in isopropanol poisoning.
Isopropanol is a central nervous system depressant that is two to three times more potent than ethanol. It is rapidly absorbed after ingestion and is metabolized by alcohol dehydrogenase to acetone, which is also a CNS depressant. Unlike other toxic alcohols, isopropanol is ketogenic but does not cause a significant metabolic acidosis. Acetone is eliminated by the lungs and kidneys. The half-life of isopropanol ranges from 3 to 16 hours, while acetone has a longer half-life of 7.5 to 26 hours. Concomitant ethanol ingestion prolongs the half-life of isopropanol but not acetone.
Isopropanol is a clear, colorless, volatile liquid with a faint acetone odor and bitter taste, commonly found as 70% rubbing alcohol. It is present in many household and industrial products including disinfectants, hand sanitizers, solvents, window cleaners, paint removers, detergents, antifreeze, and toiletries. The typical adult patient is a chronic alcoholic who ingests isopropanol after depleting ethanol supplies. Dermal, inhalational, and rectal exposures can also cause systemic toxicity. Accidental ingestions are common in young children.
Symptoms usually develop within 30 to 60 minutes of exposure. Neurologic manifestations include lethargy, inebriation, vertigo, ataxia, headache, weakness, respiratory depression, coma, and apnea, without the initial excitatory phase seen with ethanol. Gastrointestinal findings include nausea, vomiting, abdominal pain, gastritis, and hematemesis. Cardiovascular effects include hypotension, tachycardia, myocardial depression, and peripheral vasodilation. Pulmonary findings include respiratory depression and hemorrhagic tracheobronchitis. Skin and eye exposure may cause irritation or chemical burns.
Diagnosis is based on history of exposure and the characteristic odor of isopropanol or acetone on the breath. Laboratory evaluation typically shows ketosis without significant acidosis unless end-organ hypoperfusion is present. Hypoglycemia may occur, particularly in children. Acetone can falsely elevate serum creatinine, which normalizes as acetone is metabolized. Urinalysis shows ketones, and an elevated osmolar gap is common. Coma is associated with serum isopropanol levels greater than 150 mg/dL. Imaging is guided by clinical findings, with chest radiography used to assess for aspiration and head CT considered for possible trauma.
Management is primarily supportive, with attention to airway protection, breathing, and circulation. Hypotension is treated with intravenous fluids and vasopressors if needed. There is no specific antidote, and treatment with ethanol or fomepizole is contraindicated. Activated charcoal may be considered if coingestants are suspected. Skin and eye exposures require copious irrigation. Hemodialysis effectively removes isopropanol and acetone and is reserved for patients with refractory hypotension, severe toxicity, or serum levels greater than 400 mg/dL.
Patients with moderate to severe toxicity, including altered mental status or hypotension, require hospital admission. Asymptomatic or mildly intoxicated patients may be observed for 2 to 4 hours and discharged if symptoms resolve. Referral for alcohol detoxification or psychiatric evaluation is recommended for intentional ingestions.
Supportive care remains the cornerstone of treatment, and a key pitfall is inappropriate use of ethanol or fomepizole, which should be avoided in isopropanol poisoning.
- Published on
Emergency and Acute Medicine – Isopropanol poisoning
Isopropanol is a central nervous system depressant that is two to three times more potent than ethanol. It is rapidly absorbed after ingestion and is metabolized by alcohol dehydrogenase to acetone, which is also a CNS depressant. Unlike other toxic alcohols, isopropanol is ketogenic but does not cause a significant metabolic acidosis. Acetone is eliminated by the lungs and kidneys. The half-life of isopropanol ranges from 3 to 16 hours, while acetone has a longer half-life of 7.5 to 26 hours. Concomitant ethanol ingestion prolongs the half-life of isopropanol but not acetone.
Isopropanol is a clear, colorless, volatile liquid with a faint acetone odor and bitter taste, commonly found as 70% rubbing alcohol. It is present in many household and industrial products including disinfectants, hand sanitizers, solvents, window cleaners, paint removers, detergents, antifreeze, and toiletries. The typical adult patient is a chronic alcoholic who ingests isopropanol after depleting ethanol supplies. Dermal, inhalational, and rectal exposures can also cause systemic toxicity. Accidental ingestions are common in young children.
Symptoms usually develop within 30 to 60 minutes of exposure. Neurologic manifestations include lethargy, inebriation, vertigo, ataxia, headache, weakness, respiratory depression, coma, and apnea, without the initial excitatory phase seen with ethanol. Gastrointestinal findings include nausea, vomiting, abdominal pain, gastritis, and hematemesis. Cardiovascular effects include hypotension, tachycardia, myocardial depression, and peripheral vasodilation. Pulmonary findings include respiratory depression and hemorrhagic tracheobronchitis. Skin and eye exposure may cause irritation or chemical burns.
Diagnosis is based on history of exposure and the characteristic odor of isopropanol or acetone on the breath. Laboratory evaluation typically shows ketosis without significant acidosis unless end-organ hypoperfusion is present. Hypoglycemia may occur, particularly in children. Acetone can falsely elevate serum creatinine, which normalizes as acetone is metabolized. Urinalysis shows ketones, and an elevated osmolar gap is common. Coma is associated with serum isopropanol levels greater than 150 mg/dL. Imaging is guided by clinical findings, with chest radiography used to assess for aspiration and head CT considered for possible trauma.
Management is primarily supportive, with attention to airway protection, breathing, and circulation. Hypotension is treated with intravenous fluids and vasopressors if needed. There is no specific antidote, and treatment with ethanol or fomepizole is contraindicated. Activated charcoal may be considered if coingestants are suspected. Skin and eye exposures require copious irrigation. Hemodialysis effectively removes isopropanol and acetone and is reserved for patients with refractory hypotension, severe toxicity, or serum levels greater than 400 mg/dL.
Patients with moderate to severe toxicity, including altered mental status or hypotension, require hospital admission. Asymptomatic or mildly intoxicated patients may be observed for 2 to 4 hours and discharged if symptoms resolve. Referral for alcohol detoxification or psychiatric evaluation is recommended for intentional ingestions.
Supportive care remains the cornerstone of treatment, and a key pitfall is inappropriate use of ethanol or fomepizole, which should be avoided in isopropanol poisoning.
Isopropanol is a central nervous system depressant that is two to three times more potent than ethanol. It is rapidly absorbed after ingestion and is metabolized by alcohol dehydrogenase to acetone, which is also a CNS depressant. Unlike other toxic alcohols, isopropanol is ketogenic but does not cause a significant metabolic acidosis. Acetone is eliminated by the lungs and kidneys. The half-life of isopropanol ranges from 3 to 16 hours, while acetone has a longer half-life of 7.5 to 26 hours. Concomitant ethanol ingestion prolongs the half-life of isopropanol but not acetone.
Isopropanol is a clear, colorless, volatile liquid with a faint acetone odor and bitter taste, commonly found as 70% rubbing alcohol. It is present in many household and industrial products including disinfectants, hand sanitizers, solvents, window cleaners, paint removers, detergents, antifreeze, and toiletries. The typical adult patient is a chronic alcoholic who ingests isopropanol after depleting ethanol supplies. Dermal, inhalational, and rectal exposures can also cause systemic toxicity. Accidental ingestions are common in young children.
Symptoms usually develop within 30 to 60 minutes of exposure. Neurologic manifestations include lethargy, inebriation, vertigo, ataxia, headache, weakness, respiratory depression, coma, and apnea, without the initial excitatory phase seen with ethanol. Gastrointestinal findings include nausea, vomiting, abdominal pain, gastritis, and hematemesis. Cardiovascular effects include hypotension, tachycardia, myocardial depression, and peripheral vasodilation. Pulmonary findings include respiratory depression and hemorrhagic tracheobronchitis. Skin and eye exposure may cause irritation or chemical burns.
Diagnosis is based on history of exposure and the characteristic odor of isopropanol or acetone on the breath. Laboratory evaluation typically shows ketosis without significant acidosis unless end-organ hypoperfusion is present. Hypoglycemia may occur, particularly in children. Acetone can falsely elevate serum creatinine, which normalizes as acetone is metabolized. Urinalysis shows ketones, and an elevated osmolar gap is common. Coma is associated with serum isopropanol levels greater than 150 mg/dL. Imaging is guided by clinical findings, with chest radiography used to assess for aspiration and head CT considered for possible trauma.
Management is primarily supportive, with attention to airway protection, breathing, and circulation. Hypotension is treated with intravenous fluids and vasopressors if needed. There is no specific antidote, and treatment with ethanol or fomepizole is contraindicated. Activated charcoal may be considered if coingestants are suspected. Skin and eye exposures require copious irrigation. Hemodialysis effectively removes isopropanol and acetone and is reserved for patients with refractory hypotension, severe toxicity, or serum levels greater than 400 mg/dL.
Patients with moderate to severe toxicity, including altered mental status or hypotension, require hospital admission. Asymptomatic or mildly intoxicated patients may be observed for 2 to 4 hours and discharged if symptoms resolve. Referral for alcohol detoxification or psychiatric evaluation is recommended for intentional ingestions.
Supportive care remains the cornerstone of treatment, and a key pitfall is inappropriate use of ethanol or fomepizole, which should be avoided in isopropanol poisoning.
- Published on
Emergency and Acute Medicine – Isoniazid poisoning
Isoniazid (INH) poisoning results from disruption of normal vitamin B6–dependent neurotransmitter metabolism. INH complexes with and inactivates pyridoxal-5-phosphate, the active form of pyridoxine, and inhibits pyridoxine phosphokinase, preventing conversion of pyridoxine to its active form. This leads to decreased production of γ-aminobutyric acid (GABA), causing cerebral excitability and refractory seizures. INH also inhibits lactate dehydrogenase, impairing conversion of lactate to pyruvate and contributing to profound anion gap metabolic acidosis. Chronic toxicity interferes with niacin synthesis and may cause a pellagra-like syndrome after months of therapy. Rare neuropsychiatric effects include mania, depression, obsessive–compulsive symptoms, and psychosis. INH is rapidly absorbed, with peak levels in 1–2 hours, has low protein binding, and is renally excreted after hepatic acetylation. Toxicity is more severe in slow acetylators.
High-risk populations include immigrants, homeless individuals, patients with HIV, alcohol use disorder, and those of lower socioeconomic status. Slow acetylators are more prone to chronic toxicity. Ingestions less than 1.5 g usually cause mild toxicity, whereas ingestions of 10 g or more are often fatal.
Acute toxicity typically presents with neurologic findings, most notably seizures that are refractory to standard anticonvulsant therapy, along with altered mental status, agitation, coma, ataxia, hyperreflexia, hallucinations, and psychosis. Gastrointestinal symptoms include nausea and vomiting. Cardiovascular manifestations include hypotension, tachycardia, shock, and cyanosis. Metabolic findings are dominated by severe anion gap metabolic acidosis with elevated lactate and hyperthermia. Chronic toxicity presents with peripheral neuropathy, optic neuritis or atrophy, vertigo, psychosis, insomnia, and gastrointestinal or hepatic injury including hepatitis and liver failure.
Initial evaluation should focus on any patient with unexplained refractory seizures, altered mental status, or severe metabolic acidosis. Laboratory studies typically show profound metabolic acidosis on arterial blood gas, elevated anion gap, hyperglycemia, and leukocytosis in acute toxicity. Chronic toxicity may show agranulocytosis, anemia, hemolysis, and eosinophilia. Imaging is guided by clinical presentation, with chest radiography sometimes suggesting underlying tuberculosis and helping evaluate aspiration.
Management begins with airway, breathing, and circulation stabilization, supplemental oxygen, IV access, cardiac monitoring, and bedside glucose assessment. The specific antidote is pyridoxine (vitamin B6). The recommended dose is 1 g of pyridoxine for every gram of INH ingested; if the amount ingested is unknown, administer 5 g IV. Pyridoxine may be repeated if seizures or coma persist. If IV pyridoxine is unavailable, crushed tablets may be given via nasogastric tube. Benzodiazepines are used adjunctively for seizure control, but phenytoin is ineffective. Gastric decontamination with activated charcoal may be considered after stabilization, and gastric lavage is reserved for life-threatening ingestions presenting within one hour with a protected airway. Hemodialysis may be considered for persistent symptoms or renal failure. Acidosis generally resolves once seizures are controlled and does not usually require aggressive bicarbonate therapy.
Patients with refractory seizures, severe acidosis, coma, or unclear ingestion history require ICU admission. Asymptomatic patients may be discharged after a minimum of six hours of observation. Psychiatric evaluation is required for intentional ingestions.
Key pitfalls include failure to recognize INH poisoning in patients with seizures refractory to standard therapy and severe lactic acidosis. Pyridoxine should be given gram for gram relative to the suspected ingestion, and additional doses may be required if coma persists. Never paralyze a seizing patient without continuous EEG monitoring.
Isoniazid (INH) poisoning results from disruption of normal vitamin B6–dependent neurotransmitter metabolism. INH complexes with and inactivates pyridoxal-5-phosphate, the active form of pyridoxine, and inhibits pyridoxine phosphokinase, preventing conversion of pyridoxine to its active form. This leads to decreased production of γ-aminobutyric acid (GABA), causing cerebral excitability and refractory seizures. INH also inhibits lactate dehydrogenase, impairing conversion of lactate to pyruvate and contributing to profound anion gap metabolic acidosis. Chronic toxicity interferes with niacin synthesis and may cause a pellagra-like syndrome after months of therapy. Rare neuropsychiatric effects include mania, depression, obsessive–compulsive symptoms, and psychosis. INH is rapidly absorbed, with peak levels in 1–2 hours, has low protein binding, and is renally excreted after hepatic acetylation. Toxicity is more severe in slow acetylators.
High-risk populations include immigrants, homeless individuals, patients with HIV, alcohol use disorder, and those of lower socioeconomic status. Slow acetylators are more prone to chronic toxicity. Ingestions less than 1.5 g usually cause mild toxicity, whereas ingestions of 10 g or more are often fatal.
Acute toxicity typically presents with neurologic findings, most notably seizures that are refractory to standard anticonvulsant therapy, along with altered mental status, agitation, coma, ataxia, hyperreflexia, hallucinations, and psychosis. Gastrointestinal symptoms include nausea and vomiting. Cardiovascular manifestations include hypotension, tachycardia, shock, and cyanosis. Metabolic findings are dominated by severe anion gap metabolic acidosis with elevated lactate and hyperthermia. Chronic toxicity presents with peripheral neuropathy, optic neuritis or atrophy, vertigo, psychosis, insomnia, and gastrointestinal or hepatic injury including hepatitis and liver failure.
Initial evaluation should focus on any patient with unexplained refractory seizures, altered mental status, or severe metabolic acidosis. Laboratory studies typically show profound metabolic acidosis on arterial blood gas, elevated anion gap, hyperglycemia, and leukocytosis in acute toxicity. Chronic toxicity may show agranulocytosis, anemia, hemolysis, and eosinophilia. Imaging is guided by clinical presentation, with chest radiography sometimes suggesting underlying tuberculosis and helping evaluate aspiration.
Management begins with airway, breathing, and circulation stabilization, supplemental oxygen, IV access, cardiac monitoring, and bedside glucose assessment. The specific antidote is pyridoxine (vitamin B6). The recommended dose is 1 g of pyridoxine for every gram of INH ingested; if the amount ingested is unknown, administer 5 g IV. Pyridoxine may be repeated if seizures or coma persist. If IV pyridoxine is unavailable, crushed tablets may be given via nasogastric tube. Benzodiazepines are used adjunctively for seizure control, but phenytoin is ineffective. Gastric decontamination with activated charcoal may be considered after stabilization, and gastric lavage is reserved for life-threatening ingestions presenting within one hour with a protected airway. Hemodialysis may be considered for persistent symptoms or renal failure. Acidosis generally resolves once seizures are controlled and does not usually require aggressive bicarbonate therapy.
Patients with refractory seizures, severe acidosis, coma, or unclear ingestion history require ICU admission. Asymptomatic patients may be discharged after a minimum of six hours of observation. Psychiatric evaluation is required for intentional ingestions.
Key pitfalls include failure to recognize INH poisoning in patients with seizures refractory to standard therapy and severe lactic acidosis. Pyridoxine should be given gram for gram relative to the suspected ingestion, and additional doses may be required if coma persists. Never paralyze a seizing patient without continuous EEG monitoring.
- Published on
Emergency and Acute Medicine – Irritable infant
Irritability in infants is common and often part of normal development. Most infants have a predictable period of increased fussiness, usually in the evening. Normal crying peaks around 6 weeks of age, with infants crying 1–4 hours per day on average, and gradually improves over the first 6 months of life. Irritability is defined relative to the infant’s usual behavior. Colic is the most common cause of inconsolable crying, affecting up to 25% of otherwise healthy infants. It typically begins at 2–3 weeks of age, may last until 12 weeks, and is characterized by paroxysms of intense crying with knee flexion and passage of flatus. Colic is a diagnosis of exclusion.
The differential diagnosis of an irritable infant is broad and includes both benign and life-threatening conditions. Causes include gastrointestinal disorders such as gastroenteritis, gastroesophageal reflux, cow’s milk protein intolerance, constipation, anal fissure, volvulus, malrotation, intussusception, and appendicitis. Genitourinary causes include urinary tract infection, testicular torsion, incarcerated hernia, urinary retention, and genital tourniquets. Neurologic causes include increased intracranial pressure from hemorrhage, hydrocephalus, mass lesions, subdural or epidural hematomas, skull fracture, and meningitis. Cardiovascular causes include supraventricular tachycardia, congestive heart failure, myocarditis, endocarditis, anomalous coronary arteries, and coarctation of the aorta. Other important considerations include trauma, child abuse, infections (otitis media, pneumonia, thrush, gingivostomatitis), metabolic or endocrine abnormalities (hypoglycemia, hypocalcemia, hypernatremia, metabolic acidosis, inborn errors of metabolism, hyperthyroidism), toxicologic exposures, medication reactions, iron deficiency, sickle cell crisis, burns, bites, corneal abrasion or foreign body, hair or fiber tourniquets, splinters, and vaccine reactions.
Evaluation begins with careful assessment of vital signs, chief complaint, and the chronology of symptoms. A complete history should include prenatal and neonatal history, feeding patterns, stooling, sleep, recent illnesses, medications, immunizations, and caregiver concerns. Physical examination must be thorough and performed with the infant completely undressed. Measurement and plotting of weight, length, and head circumference are essential, along with rectal temperature and pulse oximetry.
Diagnostic testing is guided by the history and physical examination. Laboratory studies such as CBC, urinalysis, serum chemistries, cultures, bedside glucose testing, or stool hemoccult may be indicated. Imaging may include chest radiograph for cardiopulmonary disease, skeletal survey when abuse is suspected, CT imaging directed by neurologic findings, or contrast studies such as barium enema when intussusception is suspected. Targeted procedures may include fluorescein eye examination or ECG.
Management focuses first on identifying and stabilizing any life-threatening conditions using standard airway, breathing, and circulation principles. Immediate correction of reversible causes such as hair tourniquets or splinters is essential. If serious pathology is excluded, management is supportive. Colic is treated with reassurance, soothing rhythmic activities, reduction of environmental stimulation, and parental support. Dietary interventions such as trial of soy or hydrolyzed formula may offer temporary benefit in select infants. No medication has proven consistently effective for colic, though probiotics may help some infants. Observation in the emergency department is often appropriate.
Admission is indicated for infants with life-threatening conditions or when significant parental stress raises safety concerns. Discharge may be considered when no serious cause is identified, the infant appears well, the family is functional and supported, and close follow-up is assured. Parents must feel heard and supported, and clear return precautions are essential.
A critical pitfall is failure to recognize serious underlying disease. Cardiovascular, neurologic, gastrointestinal, metabolic, genitourinary, pulmonary, toxicologic, traumatic, ophthalmologic, and abuse-related causes must be actively considered. In noncritically ill infants, a meticulous history and complete physical examination should precede extensive laboratory or radiologic testing.
Irritability in infants is common and often part of normal development. Most infants have a predictable period of increased fussiness, usually in the evening. Normal crying peaks around 6 weeks of age, with infants crying 1–4 hours per day on average, and gradually improves over the first 6 months of life. Irritability is defined relative to the infant’s usual behavior. Colic is the most common cause of inconsolable crying, affecting up to 25% of otherwise healthy infants. It typically begins at 2–3 weeks of age, may last until 12 weeks, and is characterized by paroxysms of intense crying with knee flexion and passage of flatus. Colic is a diagnosis of exclusion.
The differential diagnosis of an irritable infant is broad and includes both benign and life-threatening conditions. Causes include gastrointestinal disorders such as gastroenteritis, gastroesophageal reflux, cow’s milk protein intolerance, constipation, anal fissure, volvulus, malrotation, intussusception, and appendicitis. Genitourinary causes include urinary tract infection, testicular torsion, incarcerated hernia, urinary retention, and genital tourniquets. Neurologic causes include increased intracranial pressure from hemorrhage, hydrocephalus, mass lesions, subdural or epidural hematomas, skull fracture, and meningitis. Cardiovascular causes include supraventricular tachycardia, congestive heart failure, myocarditis, endocarditis, anomalous coronary arteries, and coarctation of the aorta. Other important considerations include trauma, child abuse, infections (otitis media, pneumonia, thrush, gingivostomatitis), metabolic or endocrine abnormalities (hypoglycemia, hypocalcemia, hypernatremia, metabolic acidosis, inborn errors of metabolism, hyperthyroidism), toxicologic exposures, medication reactions, iron deficiency, sickle cell crisis, burns, bites, corneal abrasion or foreign body, hair or fiber tourniquets, splinters, and vaccine reactions.
Evaluation begins with careful assessment of vital signs, chief complaint, and the chronology of symptoms. A complete history should include prenatal and neonatal history, feeding patterns, stooling, sleep, recent illnesses, medications, immunizations, and caregiver concerns. Physical examination must be thorough and performed with the infant completely undressed. Measurement and plotting of weight, length, and head circumference are essential, along with rectal temperature and pulse oximetry.
Diagnostic testing is guided by the history and physical examination. Laboratory studies such as CBC, urinalysis, serum chemistries, cultures, bedside glucose testing, or stool hemoccult may be indicated. Imaging may include chest radiograph for cardiopulmonary disease, skeletal survey when abuse is suspected, CT imaging directed by neurologic findings, or contrast studies such as barium enema when intussusception is suspected. Targeted procedures may include fluorescein eye examination or ECG.
Management focuses first on identifying and stabilizing any life-threatening conditions using standard airway, breathing, and circulation principles. Immediate correction of reversible causes such as hair tourniquets or splinters is essential. If serious pathology is excluded, management is supportive. Colic is treated with reassurance, soothing rhythmic activities, reduction of environmental stimulation, and parental support. Dietary interventions such as trial of soy or hydrolyzed formula may offer temporary benefit in select infants. No medication has proven consistently effective for colic, though probiotics may help some infants. Observation in the emergency department is often appropriate.
Admission is indicated for infants with life-threatening conditions or when significant parental stress raises safety concerns. Discharge may be considered when no serious cause is identified, the infant appears well, the family is functional and supported, and close follow-up is assured. Parents must feel heard and supported, and clear return precautions are essential.
A critical pitfall is failure to recognize serious underlying disease. Cardiovascular, neurologic, gastrointestinal, metabolic, genitourinary, pulmonary, toxicologic, traumatic, ophthalmologic, and abuse-related causes must be actively considered. In noncritically ill infants, a meticulous history and complete physical examination should precede extensive laboratory or radiologic testing.

- Published on
Emergency and Acute Medicine – Irritable bowel syndrome
Irritable bowel syndrome (IBS) is a functional gastrointestinal disorder characterized by chronic abdominal pain or discomfort associated with altered bowel habits, without identifiable structural or biochemical pathology to explain the symptoms. It is common, with an estimated prevalence of 10–20% of the population.
The pathophysiology of IBS is uncertain and likely multifactorial. Proposed mechanisms include altered gastrointestinal motility and increased gut sensitivity (visceral hyperalgesia), leading to exaggerated pain responses to normal bowel activity. Mucosal inflammation may play a role, particularly in postinfectious IBS, which can occur in up to 10% of patients following bacterial enteritis and is associated with mucosal lymphocyte infiltration. Altered intestinal microflora has also been described. Food sensitivity is frequently reported by patients but remains unproven as a primary cause. Psychosocial factors are important in some patients, especially those who seek medical care, with higher rates of anxiety, somatoform disorders, and prior abuse histories; however, there is no clear increase in psychiatric illness among individuals with IBS who do not seek care.
Clinically, IBS presents with recurrent abdominal pain or discomfort that is often relieved by defecation and associated with changes in stool frequency or consistency. Common accompanying symptoms include bloating or abdominal distention, passage of mucus, and a sensation of incomplete evacuation. The Rome III criteria define IBS as recurrent abdominal pain or discomfort at least 3 days per month over the past 3 months, associated with at least two of the following: improvement with defecation, onset associated with a change in stool frequency, or onset associated with a change in stool form. IBS is more common in women than men, particularly among those who seek medical attention.
A careful history is central to diagnosis, as IBS is a clinical diagnosis. Alarm features that warrant further evaluation include onset after age 50, nocturnal symptoms, unintentional weight loss, iron-deficiency anemia, hematochezia, or a family history of colorectal cancer, inflammatory bowel disease, or celiac disease. Physical examination is usually normal, though mild sigmoid tenderness or a palpable sigmoid cord may be noted.
Laboratory testing is typically normal and is not required to establish the diagnosis. When indicated to exclude alternative diagnoses, basic studies such as a CBC (to rule out anemia or leukocytosis), ESR or CRP (to exclude inflammatory disease), and selective stool studies for diarrhea may be considered. Testing for celiac disease may be appropriate in the outpatient setting. Imaging and endoscopic evaluation are reserved for patients with alarm features or diagnostic uncertainty.
Management in the emergency setting is supportive and focused on reassurance. Establishing an empathetic physician–patient relationship is essential. Treatment is individualized and symptom based. Lifestyle interventions such as regular exercise may improve constipation and overall symptoms. Dietary modification can be empirically attempted, including exclusion of lactose or gluten and avoidance of gas-producing foods if these worsen symptoms. Constipation-predominant symptoms may benefit from increased dietary fiber or fiber supplementation. Abdominal pain may improve with short-term use of antispasmodics such as dicyclomine or hyoscyamine. Probiotics, particularly those containing bifidobacteria, may provide benefit. Low-dose tricyclic antidepressants or selective serotonin reuptake inhibitors can reduce global IBS symptoms and abdominal pain, and psychological therapies are effective in selected patients.
Most patients with IBS can be safely discharged from the emergency department with reassurance and outpatient follow-up. Admission is rarely indicated and should be reserved for patients with diagnostic uncertainty or concern for emergent abdominal pathology. Ongoing care with a primary care physician is critical, and some patients may benefit from gastroenterology or mental health referral.
A key pitfall is prematurely attributing symptoms to IBS without adequately considering alternative or emergent diagnoses. Although IBS is common and often underlies repeated emergency evaluations, alarm features or atypical presentations should prompt further investigation.
Irritable bowel syndrome (IBS) is a functional gastrointestinal disorder characterized by chronic abdominal pain or discomfort associated with altered bowel habits, without identifiable structural or biochemical pathology to explain the symptoms. It is common, with an estimated prevalence of 10–20% of the population.
The pathophysiology of IBS is uncertain and likely multifactorial. Proposed mechanisms include altered gastrointestinal motility and increased gut sensitivity (visceral hyperalgesia), leading to exaggerated pain responses to normal bowel activity. Mucosal inflammation may play a role, particularly in postinfectious IBS, which can occur in up to 10% of patients following bacterial enteritis and is associated with mucosal lymphocyte infiltration. Altered intestinal microflora has also been described. Food sensitivity is frequently reported by patients but remains unproven as a primary cause. Psychosocial factors are important in some patients, especially those who seek medical care, with higher rates of anxiety, somatoform disorders, and prior abuse histories; however, there is no clear increase in psychiatric illness among individuals with IBS who do not seek care.
Clinically, IBS presents with recurrent abdominal pain or discomfort that is often relieved by defecation and associated with changes in stool frequency or consistency. Common accompanying symptoms include bloating or abdominal distention, passage of mucus, and a sensation of incomplete evacuation. The Rome III criteria define IBS as recurrent abdominal pain or discomfort at least 3 days per month over the past 3 months, associated with at least two of the following: improvement with defecation, onset associated with a change in stool frequency, or onset associated with a change in stool form. IBS is more common in women than men, particularly among those who seek medical attention.
A careful history is central to diagnosis, as IBS is a clinical diagnosis. Alarm features that warrant further evaluation include onset after age 50, nocturnal symptoms, unintentional weight loss, iron-deficiency anemia, hematochezia, or a family history of colorectal cancer, inflammatory bowel disease, or celiac disease. Physical examination is usually normal, though mild sigmoid tenderness or a palpable sigmoid cord may be noted.
Laboratory testing is typically normal and is not required to establish the diagnosis. When indicated to exclude alternative diagnoses, basic studies such as a CBC (to rule out anemia or leukocytosis), ESR or CRP (to exclude inflammatory disease), and selective stool studies for diarrhea may be considered. Testing for celiac disease may be appropriate in the outpatient setting. Imaging and endoscopic evaluation are reserved for patients with alarm features or diagnostic uncertainty.
Management in the emergency setting is supportive and focused on reassurance. Establishing an empathetic physician–patient relationship is essential. Treatment is individualized and symptom based. Lifestyle interventions such as regular exercise may improve constipation and overall symptoms. Dietary modification can be empirically attempted, including exclusion of lactose or gluten and avoidance of gas-producing foods if these worsen symptoms. Constipation-predominant symptoms may benefit from increased dietary fiber or fiber supplementation. Abdominal pain may improve with short-term use of antispasmodics such as dicyclomine or hyoscyamine. Probiotics, particularly those containing bifidobacteria, may provide benefit. Low-dose tricyclic antidepressants or selective serotonin reuptake inhibitors can reduce global IBS symptoms and abdominal pain, and psychological therapies are effective in selected patients.
Most patients with IBS can be safely discharged from the emergency department with reassurance and outpatient follow-up. Admission is rarely indicated and should be reserved for patients with diagnostic uncertainty or concern for emergent abdominal pathology. Ongoing care with a primary care physician is critical, and some patients may benefit from gastroenterology or mental health referral.
A key pitfall is prematurely attributing symptoms to IBS without adequately considering alternative or emergent diagnoses. Although IBS is common and often underlies repeated emergency evaluations, alarm features or atypical presentations should prompt further investigation.

- Published on
Emergency and Acute Medicine – Iron Poisoning
Basics and pathophysiology
Iron poisoning is a potentially life-threatening toxicologic emergency, most commonly seen in pediatric accidental ingestions and adult intentional overdoses. Peak serum iron concentrations usually occur 2–4 hours after ingestion. Serum iron levels obtained more than 4–6 hours after ingestion may be misleading because iron rapidly redistributes into tissues; enteric-coated or sustained-release preparations require serial measurements. After absorption, free iron shifts intracellularly, causing direct cellular injury. The gastrointestinal tract sustains corrosive injury that can lead to hemorrhage, profound fluid loss, shock, and perforation. The liver receives the highest iron load via portal circulation, resulting in hemorrhagic periportal necrosis. Free iron disrupts mitochondrial oxidative phosphorylation, catalyzes lipid peroxidation and free radical formation, increases anaerobic metabolism, and produces metabolic acidosis. Cardiovascular depression, venodilation, cerebral edema, and acidemia result from these mechanisms.
Etiology and toxic doses
Toxicity depends on the amount of elemental iron ingested. Doses below 20 mg/kg are generally nontoxic, while ingestions greater than 40 mg/kg are associated with moderate to severe toxicity. Lethality is possible at doses exceeding 60 mg/kg. Elemental iron content varies by formulation: ferrous sulfate contains approximately 20% elemental iron (a 325-mg tablet contains 65 mg elemental iron), ferrous gluconate contains 12%, and ferrous fumarate contains 33%. Prenatal vitamins may contain 60–90 mg elemental iron per tablet, whereas children’s vitamins usually contain 5–18 mg per tablet. Historically, iron poisoning carried one of the highest pediatric mortality rates due to adult iron products; modern children’s chewable preparations are considerably safer.
Clinical presentation
Iron poisoning classically progresses through five stages, though patients may skip stages or present at any point. Stage 1 (0.5–6 hours) is characterized by gastrointestinal symptoms including abdominal pain, vomiting, diarrhea, hematemesis, and hematochezia. Stage 2 (6–24 hours) is a deceptive latent phase during which GI symptoms improve despite ongoing cellular injury; hypotension and acidosis may develop. Stage 3 (6–72 hours) involves shock and systemic toxicity with hypoperfusion, severe metabolic acidosis, coma, and coagulopathy. Stage 4 (2–3 days) is marked by hepatic failure with hypoglycemia, jaundice, coagulopathy, and markedly elevated transaminases. Stage 5 (2–4 weeks) presents with gastric outlet or small bowel obstruction due to scarring. Absence of symptoms within the first 6 hours makes a significant ingestion unlikely.
Diagnosis and essential workup
Acute iron poisoning is primarily a clinical diagnosis, and treatment decisions should not rely solely on laboratory values. Serum iron levels peak between 2 and 6 hours after ingestion, with 4 hours being the most common peak; delayed peaks occur with sustained-release formulations. Laboratory evaluation typically reveals an anion gap metabolic acidosis, early hyperglycemia followed by late hypoglycemia, leukocytosis, anemia if bleeding is significant, abnormal liver function tests, and coagulopathy in severe cases. Total iron-binding capacity is not useful and should not be obtained. Abdominal radiographs may reveal radiopaque tablets, although absence of pill fragments does not exclude severe poisoning. Imaging is also useful for detecting perforation.
Differential diagnosis
The differential diagnosis includes sepsis, acetaminophen toxicity, gastrointestinal bleeding from other causes, and toxic ingestions that cause anion gap metabolic acidosis such as salicylates, cyanide, methanol, ethylene glycol, heavy metals, mushrooms, and theophylline.
Emergency management
Initial management focuses on airway, breathing, and circulation. Patients with altered mental status or hemodynamic instability may require intubation. Establish venous access, initiate aggressive fluid resuscitation for hypotension, and place the patient on cardiac monitoring and pulse oximetry. Activated charcoal is ineffective for iron and should not be used. Gastric lavage has not shown outcome benefit. Sodium bicarbonate, phosphate preparations, and oral deferoxamine are not recommended. If tablets are visualized on abdominal radiograph or there is a history of significant ingestion, whole bowel irrigation with polyethylene glycol solution via nasogastric tube may be considered, with careful monitoring for gastrointestinal bleeding or perforation. Endoscopic or surgical removal may be required for massive ingestions with bezoar formation.
Chelation therapy
Deferoxamine (DFO) is the treatment of choice for significant iron poisoning and should be administered as early as possible, ideally within 24 hours. It chelates free iron, forming a complex excreted in urine, which may turn a characteristic “vin rose” color. Intravenous infusion is preferred and should be titrated carefully due to the risk of hypotension; infusion rates may be increased to 15 mg/kg/hour and adjusted as tolerated. Treatment decisions should be based on clinical status as well as laboratory data, especially in late presenters, because serum iron levels may be deceptively low after redistribution. Prolonged DFO therapy beyond 24–48 hours increases the risk of acute respiratory distress syndrome and should be used cautiously. Resolution of acidosis and systemic toxicity is the best endpoint for therapy. Regional poison control consultation is strongly recommended for moderate to severe cases.
Disposition and follow-up
Patients with gastrointestinal symptoms, dehydration, altered mental status, hypotension, metabolic acidosis, shock, serum iron levels above 500 mg/dL, rising iron levels, or those requiring deferoxamine therapy should be admitted. ICU admission is indicated for coma, severe acidosis, shock, or extremely elevated iron levels. Patients who remain asymptomatic after 6 hours of observation, have normal radiographs, minimal symptoms, and serum iron levels below 350 mg/dL may be discharged with careful instructions. Follow-up is important for patients at risk of delayed gastric outlet obstruction. Psychiatric evaluation is required for intentional ingestions.
Pearls and pitfalls
Resolution of early gastrointestinal symptoms does not exclude ongoing or worsening toxicity. Deferoxamine may be indicated even in late presentations with relatively low serum iron levels if there is evidence of intracellular poisoning such as metabolic acidosis. Clinical status, not laboratory values alone, should guide management decisions.
Basics and pathophysiology
Iron poisoning is a potentially life-threatening toxicologic emergency, most commonly seen in pediatric accidental ingestions and adult intentional overdoses. Peak serum iron concentrations usually occur 2–4 hours after ingestion. Serum iron levels obtained more than 4–6 hours after ingestion may be misleading because iron rapidly redistributes into tissues; enteric-coated or sustained-release preparations require serial measurements. After absorption, free iron shifts intracellularly, causing direct cellular injury. The gastrointestinal tract sustains corrosive injury that can lead to hemorrhage, profound fluid loss, shock, and perforation. The liver receives the highest iron load via portal circulation, resulting in hemorrhagic periportal necrosis. Free iron disrupts mitochondrial oxidative phosphorylation, catalyzes lipid peroxidation and free radical formation, increases anaerobic metabolism, and produces metabolic acidosis. Cardiovascular depression, venodilation, cerebral edema, and acidemia result from these mechanisms.
Etiology and toxic doses
Toxicity depends on the amount of elemental iron ingested. Doses below 20 mg/kg are generally nontoxic, while ingestions greater than 40 mg/kg are associated with moderate to severe toxicity. Lethality is possible at doses exceeding 60 mg/kg. Elemental iron content varies by formulation: ferrous sulfate contains approximately 20% elemental iron (a 325-mg tablet contains 65 mg elemental iron), ferrous gluconate contains 12%, and ferrous fumarate contains 33%. Prenatal vitamins may contain 60–90 mg elemental iron per tablet, whereas children’s vitamins usually contain 5–18 mg per tablet. Historically, iron poisoning carried one of the highest pediatric mortality rates due to adult iron products; modern children’s chewable preparations are considerably safer.
Clinical presentation
Iron poisoning classically progresses through five stages, though patients may skip stages or present at any point. Stage 1 (0.5–6 hours) is characterized by gastrointestinal symptoms including abdominal pain, vomiting, diarrhea, hematemesis, and hematochezia. Stage 2 (6–24 hours) is a deceptive latent phase during which GI symptoms improve despite ongoing cellular injury; hypotension and acidosis may develop. Stage 3 (6–72 hours) involves shock and systemic toxicity with hypoperfusion, severe metabolic acidosis, coma, and coagulopathy. Stage 4 (2–3 days) is marked by hepatic failure with hypoglycemia, jaundice, coagulopathy, and markedly elevated transaminases. Stage 5 (2–4 weeks) presents with gastric outlet or small bowel obstruction due to scarring. Absence of symptoms within the first 6 hours makes a significant ingestion unlikely.
Diagnosis and essential workup
Acute iron poisoning is primarily a clinical diagnosis, and treatment decisions should not rely solely on laboratory values. Serum iron levels peak between 2 and 6 hours after ingestion, with 4 hours being the most common peak; delayed peaks occur with sustained-release formulations. Laboratory evaluation typically reveals an anion gap metabolic acidosis, early hyperglycemia followed by late hypoglycemia, leukocytosis, anemia if bleeding is significant, abnormal liver function tests, and coagulopathy in severe cases. Total iron-binding capacity is not useful and should not be obtained. Abdominal radiographs may reveal radiopaque tablets, although absence of pill fragments does not exclude severe poisoning. Imaging is also useful for detecting perforation.
Differential diagnosis
The differential diagnosis includes sepsis, acetaminophen toxicity, gastrointestinal bleeding from other causes, and toxic ingestions that cause anion gap metabolic acidosis such as salicylates, cyanide, methanol, ethylene glycol, heavy metals, mushrooms, and theophylline.
Emergency management
Initial management focuses on airway, breathing, and circulation. Patients with altered mental status or hemodynamic instability may require intubation. Establish venous access, initiate aggressive fluid resuscitation for hypotension, and place the patient on cardiac monitoring and pulse oximetry. Activated charcoal is ineffective for iron and should not be used. Gastric lavage has not shown outcome benefit. Sodium bicarbonate, phosphate preparations, and oral deferoxamine are not recommended. If tablets are visualized on abdominal radiograph or there is a history of significant ingestion, whole bowel irrigation with polyethylene glycol solution via nasogastric tube may be considered, with careful monitoring for gastrointestinal bleeding or perforation. Endoscopic or surgical removal may be required for massive ingestions with bezoar formation.
Chelation therapy
Deferoxamine (DFO) is the treatment of choice for significant iron poisoning and should be administered as early as possible, ideally within 24 hours. It chelates free iron, forming a complex excreted in urine, which may turn a characteristic “vin rose” color. Intravenous infusion is preferred and should be titrated carefully due to the risk of hypotension; infusion rates may be increased to 15 mg/kg/hour and adjusted as tolerated. Treatment decisions should be based on clinical status as well as laboratory data, especially in late presenters, because serum iron levels may be deceptively low after redistribution. Prolonged DFO therapy beyond 24–48 hours increases the risk of acute respiratory distress syndrome and should be used cautiously. Resolution of acidosis and systemic toxicity is the best endpoint for therapy. Regional poison control consultation is strongly recommended for moderate to severe cases.
Disposition and follow-up
Patients with gastrointestinal symptoms, dehydration, altered mental status, hypotension, metabolic acidosis, shock, serum iron levels above 500 mg/dL, rising iron levels, or those requiring deferoxamine therapy should be admitted. ICU admission is indicated for coma, severe acidosis, shock, or extremely elevated iron levels. Patients who remain asymptomatic after 6 hours of observation, have normal radiographs, minimal symptoms, and serum iron levels below 350 mg/dL may be discharged with careful instructions. Follow-up is important for patients at risk of delayed gastric outlet obstruction. Psychiatric evaluation is required for intentional ingestions.
Pearls and pitfalls
Resolution of early gastrointestinal symptoms does not exclude ongoing or worsening toxicity. Deferoxamine may be indicated even in late presentations with relatively low serum iron levels if there is evidence of intracellular poisoning such as metabolic acidosis. Clinical status, not laboratory values alone, should guide management decisions.

