- Published on
Pathology - Malignant melanoma
A malignant melanocytic tumor.
Epidemiology • Less prevalent than basal or squamous cell carcinomas of the skin, however far more lethal. • Primarily observed in those with pale skin who have been exposed to sunlight.
Aetiology • Intermittent exposure to high doses of UV radiation constitutes the primary risk factor. • A component of genetic predisposition may also be pertinent. Genetics • Melanomas that develop in areas intermittently exposed to sunlight generally exhibit mutations in BRAF as an initial genetic occurrence. • Progression correlates with the accumulation of mutations in genes such as KIT, MITF, CDKN2A, TP53, and PTEN. • The majority also have chromosomal abnormalities characterized by gains and/or losses of chromosomal segments.
Presentation • The majority of melanomas manifest as pigmented cutaneous lesions exhibiting A symmetry, irregular B ordering, heterogeneous C olour, and D iameter exceeding 6mm (the ‘ABCD’ abbreviation).
Histopathology • A characteristic feature of all types of malignant melanoma is the existence of a neoplastic proliferation of markedly abnormal melanocytes. If the process is restricted to the epidermis, the term melanoma in situ may be utilized. • The term invasive melanoma may be utilized once penetration into the dermis has transpired.
Evolution • The majority of melanomas initially develop as a flat lesion in a radial manner, referred to as the radial growth phase. In this phase, there is either an absence of dermal invasion or the cells within the dermis are incapable of surviving and proliferating. • As advancement occurs, the growth transitions, enabling cells inside the dermis to proliferate. This phase is referred to as the vertical growth phase and is linked to the development of metastatic potential
Prognosis: Survival correlates with the disease stage at the time of diagnosis. • The primary factors determining the stage are the Breslow thickness of the melanoma and the presence of ulceration. • The mitotic rate is also acknowledged as a significant prognostic predictor in vertical growth phase melanomas.
A malignant melanocytic tumor.
Epidemiology • Less prevalent than basal or squamous cell carcinomas of the skin, however far more lethal. • Primarily observed in those with pale skin who have been exposed to sunlight.
Aetiology • Intermittent exposure to high doses of UV radiation constitutes the primary risk factor. • A component of genetic predisposition may also be pertinent. Genetics • Melanomas that develop in areas intermittently exposed to sunlight generally exhibit mutations in BRAF as an initial genetic occurrence. • Progression correlates with the accumulation of mutations in genes such as KIT, MITF, CDKN2A, TP53, and PTEN. • The majority also have chromosomal abnormalities characterized by gains and/or losses of chromosomal segments.
Presentation • The majority of melanomas manifest as pigmented cutaneous lesions exhibiting A symmetry, irregular B ordering, heterogeneous C olour, and D iameter exceeding 6mm (the ‘ABCD’ abbreviation).
Histopathology • A characteristic feature of all types of malignant melanoma is the existence of a neoplastic proliferation of markedly abnormal melanocytes. If the process is restricted to the epidermis, the term melanoma in situ may be utilized. • The term invasive melanoma may be utilized once penetration into the dermis has transpired.
Evolution • The majority of melanomas initially develop as a flat lesion in a radial manner, referred to as the radial growth phase. In this phase, there is either an absence of dermal invasion or the cells within the dermis are incapable of surviving and proliferating. • As advancement occurs, the growth transitions, enabling cells inside the dermis to proliferate. This phase is referred to as the vertical growth phase and is linked to the development of metastatic potential
Prognosis: Survival correlates with the disease stage at the time of diagnosis. • The primary factors determining the stage are the Breslow thickness of the melanoma and the presence of ulceration. • The mitotic rate is also acknowledged as a significant prognostic predictor in vertical growth phase melanomas.
- Published on
Pathology - Mycosis fungoides
A low-grade T-cell lymphoma characterized by variably epidermotropic skin-homing T-lymphocytes.
Epidemiology: The most prevalent type of primary cutaneous lymphoma, however it remains a rare condition, impacting 0.3 individuals per 100,000 yearly. • Typically an affliction of maturity, however it may sporadically impact children.
Aetiology: Unknown. Genetics • The course of the disease correlates with chromosomal abnormalities, especially those involving chromosomes 8 and 17.
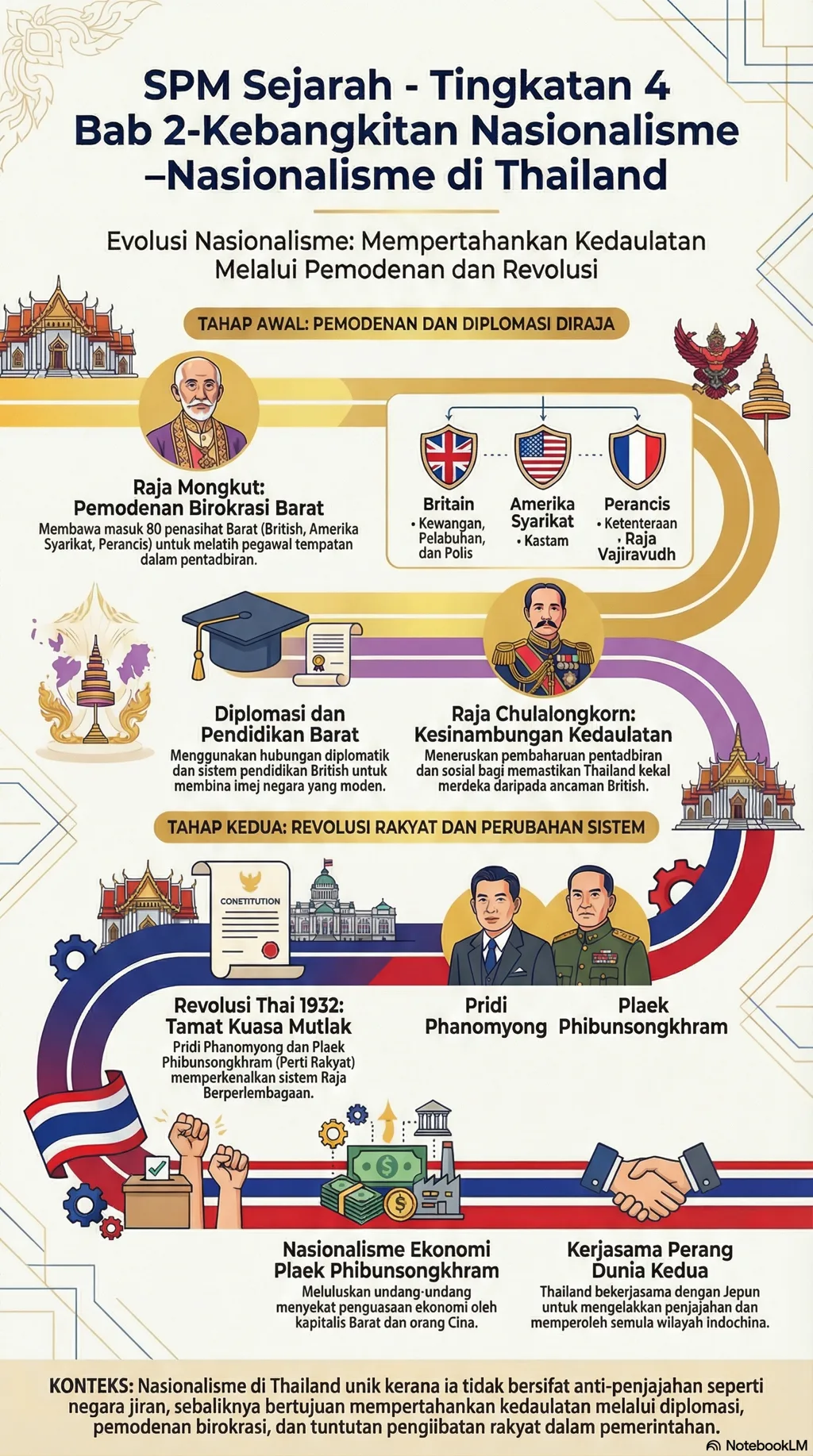
Presentation • Defined by the progressive emergence of patches, plaques, and tumors on non-sun-exposed skin, mainly in the regions of the buttocks and trunk.
Patches are several extensive (> 10mm) flat erythematous scaly sores.
• Plaques are raised lesions that may arise inside existing patches or develop independently.
• Tumour nodules and, at times, erythroderma may subsequently arise. • Advanced illness may affect the bone marrow, lymph nodes, and visceral organs.
Histopathology The patch stage has a modest upper dermal T-cell infiltrate accompanied by variable epidermotropism. The early stages of the disease are frequently challenging to detect due to overlapping characteristics with many inflammatory disorders. • The plaque stage has a more pronounced, band-like infiltration of T-cells with increased epidermotropism. Aggregates of neoplastic cells within the epidermis are commonly observed (Pautrier microabscesses). The nuclear atypia of the lymphocytes is more pronounced. The tumor stage indicates a more widespread skin infiltrate that may migrate into the subcutaneous fat. Epidermotropism may be diminished. Immunophenotype: Most instances exhibit a T-helper cell phenotype, specifically CD3 + CD4 + CD8–.
Prognosis: The likelihood of progression and mortality is associated with the illness stage at presentation. • The 10-year survival rates are elevated (85–95%) in patch and plaque stage disease, decreasing to 40% in tumor stage, and to 20% with nodal involvement.
A low-grade T-cell lymphoma characterized by variably epidermotropic skin-homing T-lymphocytes.
Epidemiology: The most prevalent type of primary cutaneous lymphoma, however it remains a rare condition, impacting 0.3 individuals per 100,000 yearly. • Typically an affliction of maturity, however it may sporadically impact children.
Aetiology: Unknown. Genetics • The course of the disease correlates with chromosomal abnormalities, especially those involving chromosomes 8 and 17.
Presentation • Defined by the progressive emergence of patches, plaques, and tumors on non-sun-exposed skin, mainly in the regions of the buttocks and trunk.
Patches are several extensive (> 10mm) flat erythematous scaly sores.
• Plaques are raised lesions that may arise inside existing patches or develop independently.
• Tumour nodules and, at times, erythroderma may subsequently arise. • Advanced illness may affect the bone marrow, lymph nodes, and visceral organs.
Histopathology The patch stage has a modest upper dermal T-cell infiltrate accompanied by variable epidermotropism. The early stages of the disease are frequently challenging to detect due to overlapping characteristics with many inflammatory disorders. • The plaque stage has a more pronounced, band-like infiltration of T-cells with increased epidermotropism. Aggregates of neoplastic cells within the epidermis are commonly observed (Pautrier microabscesses). The nuclear atypia of the lymphocytes is more pronounced. The tumor stage indicates a more widespread skin infiltrate that may migrate into the subcutaneous fat. Epidermotropism may be diminished. Immunophenotype: Most instances exhibit a T-helper cell phenotype, specifically CD3 + CD4 + CD8–.
Prognosis: The likelihood of progression and mortality is associated with the illness stage at presentation. • The 10-year survival rates are elevated (85–95%) in patch and plaque stage disease, decreasing to 40% in tumor stage, and to 20% with nodal involvement.

- Published on
Pathology – Benign Bone Tumors
I. Osteochondroma
I. Osteochondroma
- Definition: Benign bone-forming tumour.
- Location: Solitary exophytic nodule arising from the metaphysis of long bones near the epiphyseal growth plate.
- Population: Common in children.
- Histology:
- Outer fibrous perichondrium.
- Cartilage cap.
- Underlying bony stalk.
- Key takeaway: Think "bone spur" growing outwards from the growth plate of a long bone in a child.
- Definition: Benign cartilage-forming tumour.
- Location: Two main types:
- Enchondromas: Medulla of bones (hands and feet are common).
- Periosteal chondromas: Surface of bone (proximal humerus is characteristic).
- Presentation: Often discovered incidentally (no symptoms).
- Histology: Chondrocytes within an abundant cartilaginous matrix.
- Key takeaway: Cartilage-based tumour, location distinguishes the subtypes.
- Definition: Benign bone-forming tumour.
- Location: Long bones of children/young adults (proximal femur is common).
- Presentation: Characteristic nocturnal pain.
- Radiographic Appearance: Small lucent nidus (<1cm) on plain radiographs.< />pan>
- Histology: Well-circumscribed, hypervascular area of bony trabeculae surrounded by reactive bone.
- Key takeaway: Small, painful lesion easily visualized on X-ray; pain is a key clinical feature.
- Definition: Benign, but locally aggressive neoplasm.
- Location: Ends of long bones.
- Population: Young adults (20-45 years).
- Presentation: Pain and swelling at the tumour site.
- Histology: Sheets of neoplastic ovoid mononuclear cells with interspersed large osteoclast-like giant cells.
- Prognosis: ~25% local recurrence rate after excision. Distant metastasis is rare.
- Key takeaway: Although benign, its local aggressiveness and potential for recurrence are significant.

- Published on
- Published on
Pathology - Rheumatoid Arthritis
I. Definition & Epidemiology
RA can affect multiple organ systems:
RA has a variable course:
I. Definition & Epidemiology
- Definition: RA is a systemic autoimmune disease primarily affecting synovial joints. It's characterized by chronic inflammation.
- Epidemiology: Affects approximately 1% of the population, with a significantly higher prevalence in young to middle-aged women.
- Etiology (Cause): The initial trigger for RA remains unknown. However, once inflammation starts, it becomes self-sustaining.
- Pathogenesis (Mechanism):
- Inflammation: The synovium (joint lining) is infiltrated by CD4+ T-cells, B-cells, plasma cells, and macrophages, leading to inflammation.
- Pannus Formation: The inflamed synovium proliferates, forming pannus – a hyperplastic mass of synovial tissue.
- Joint Destruction: Pannus erodes articular cartilage, resulting in joint damage.
- Joint Involvement: Symmetrical swelling, pain, and stiffness, predominantly affecting small joints of the hands and feet. Morning stiffness is a characteristic symptom.
- Rheumatoid Factor (RF): An autoantibody targeting the Fc portion of IgG. Positive in ~70% of RA patients, but also found in other autoimmune diseases and some healthy individuals, limiting its specificity.
- Anti-citrullinated protein antibodies (ACPAs): Newer, more specific antibodies for diagnosing RA, but not as widely available as RF.
RA can affect multiple organ systems:
- Cardiovascular: Ischemic heart disease, pericarditis
- Vascular: Accelerated atherosclerosis, vasculitis
- Hematological: Anemia, splenomegaly
- Pulmonary: Pulmonary fibrosis, pleuritis
- Skin: Rheumatoid nodules, erythema nodosum, pyoderma gangrenosum
- Neurological: Peripheral neuropathy, stroke
- Amyloidosis: Deposition of serum amyloid A, leading to AA amyloidosis in various organs.
- Synovial Hyperplasia: Marked increase in synovial tissue.
- Inflammatory Infiltrate: A significant presence of lymphocytes and plasma cells.
- Germinal Centers: Formation of germinal centers within lymphoid aggregates is a typical finding.
RA has a variable course:
- Remission: Approximately 25% experience long-term remission.
- Mild-to-Moderate Disability: About 50% have chronic disease with mild-to-moderate disability.
- Severe Disability: Approximately 25% experience progressive disease leading to severe disability.
- Autoimmune nature: RA is driven by the body's immune system attacking its own tissues.
- Synovitis: Inflammation of the synovial membrane is central to the disease process.
- Pannus formation & joint destruction: Understand the sequence of events leading to joint damage.
- Diagnostic markers: Compare the sensitivity and specificity of RF and ACPAs.
- Systemic involvement: Be aware of the diverse extra-articular manifestations.
- Published on
Pathology - Osteoarthritis
I. Definition & Epidemiology:
I. Definition & Epidemiology:
- Definition: Osteoarthritis (OA) is a group of diseases causing joint degradation. Crucially, it's not a single disease but a collection of similar conditions.
- Epidemiology: OA is the most common joint disease. In the UK, approximately 2 million people experience symptomatic OA. It primarily affects the elderly population.
- Primary OA: In most cases, the cause of OA is unknown (primary OA). This highlights the complexity of the disease and the challenges in pinpointing a single cause.
- Secondary OA: OA can also develop as a consequence of other joint disorders (secondary OA). Examples include rheumatoid arthritis and gout, where pre-existing joint damage sets the stage for OA development. Understanding this distinction is vital – treatment differs depending on the underlying cause.
- Cartilage Damage: The primary pathological change is damage to the articular cartilage, the smooth tissue covering joint surfaces. This damage is central to understanding OA's progression.
- Inflammation & Metalloproteinases: Low-grade inflammation within the joint plays a crucial role. This inflammation causes chondrocytes (cartilage cells) to release metalloproteinases, enzymes that break down the cartilage matrix, leading to further cartilage loss.
- Subchondral Bone Response: As cartilage is lost, the underlying bone becomes exposed. The bone responds by thickening (sclerosis) – a compensatory but ultimately unhelpful response. This bone thickening contributes to the pain and stiffness experienced by patients.
- Pain, Tenderness, Stiffness: The classic symptoms are pain, tenderness, and stiffness in the affected joint(s). It's important to note that symptom severity varies greatly between individuals.
- Activity-Related Worsening: Symptoms typically worsen throughout the day, particularly with increased activity. This contrasts with some other inflammatory arthritides where stiffness is worse in the morning.
- Commonly Affected Joints: OA most commonly affects the hip, knee, spine, and small joints of the hands, but it can affect any joint. Knowing the typical locations helps in differential diagnosis.
- Cartilage Loss & Thinning: Microscopic examination reveals thinned and lost articular cartilage. This confirms the central role of cartilage degeneration in the disease process.
- Subchondral Bone Sclerosis: Thickening and sclerosis (increased density) of the subchondral bone are also observed. This supports the clinical observation of bone changes in OA.
- Progressive Worsening: OA generally worsens over time. This emphasizes the need for early diagnosis and management to slow progression.
- Analgesics & Joint Replacement: Management typically involves analgesics (pain relievers) to manage symptoms. In severe cases, joint replacement surgery may be necessary. This highlights the potential long-term impact of the disease.

- Published on
Pathology - Malignant Bone Tumors
I. Metastatic Bone Tumors
A. Osteosarcoma
I. Metastatic Bone Tumors
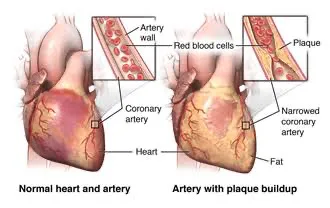
- Prevalence: The vast majority of malignant bone tumors are metastatic (secondary), meaning they spread from another primary cancer site.
- Primary Sources: Common primary sites include lung, breast, kidney, thyroid, and prostate carcinomas.
- Bone Destruction: Most metastatic bone tumors are osteolytic, meaning they break down bone tissue. An exception is prostate cancer metastases, which are typically osteosclerotic (stimulate new bone formation).
- Hypercalcemia: Bone metastases are a frequent cause of hypercalcemia (high blood calcium levels).
A. Osteosarcoma
- Definition: A malignant bone-forming tumor; the most common primary malignant bone tumor.
- Demographics: Most commonly affects individuals aged 5-25, presenting with persistent deep pain in a long bone, often accompanied by a palpable mass. Elderly cases are often secondary to Paget's disease.
- Histology: Composed of atypical cells that produce osteoid (unmineralized bone matrix).
- Spread: Highly malignant with early and rapid hematogenous (bloodstream) spread, primarily to the lungs.
- Prognosis: Strongly dependent on response to preoperative therapy. Responders have excellent survival (80-90%), while non-responders have poor survival (<15%).< />pan>
- Definition: A malignant cartilage-forming tumor; the second most common primary malignant bone tumor.
- Demographics: Primarily affects adults over 50 years old.
- Location: Most common in pelvic bones.
- Histology: Composed of variably atypical cells producing a cartilaginous matrix. Graded 1-3, with most being grades 1 or 2.
- Prognosis: Histological grade is the key prognostic factor. Grade 1 tumors have a 90% 5-year survival rate, while grades 2 and 3 have closer to 50%.
- Definition: A malignant round cell tumor of bone.
- Demographics: Most common in children and adolescents under 20 years old.
- Presentation: Presents with pain and a mass in long bones, pelvis, or ribs.
- Histology: Small round tumor cells with scant cytoplasm; may form rosette-like structures; necrosis is common.
- Genetics: Often shows a characteristic t(11;22) chromosomal translocation fusing the EWS and FLI1 genes.
- Prognosis: Survival rates are less than 50%, depending on stage, location, and tumor size.
- Published on
Pathology - Osteomalacia.
I. Definition & Epidemiology:
I. Definition & Epidemiology:
- Definition: Osteomalacia is a metabolic bone disease. Crucially, it's characterized by inadequate mineralization of osteoid, resulting in soft, weakened bones. This means the bone matrix (osteoid) doesn't harden properly.
- Epidemiology: Relatively rare in developed nations. Predominantly affects the elderly population.
- Primary Cause: Almost all cases stem from vitamin D deficiency. Understanding the why behind the deficiency is critical.
- Causes of Vitamin D Deficiency:
- Insufficient sun exposure: Lack of UV-B radiation needed for vitamin D synthesis in the skin.
- Malabsorption: Conditions affecting nutrient absorption from the gut (e.g., celiac disease, Crohn's disease).
- Chronic liver disease: Impaired vitamin D metabolism.
- Chronic kidney disease: Reduced ability to activate vitamin D in the kidneys.
- Bone Matrix Mineralization Failure: Insufficient mineralization of the bone matrix (osteoid) leads to a buildup of unmineralized osteoid. This is the hallmark of the disease.
- Structural Weakness: The excess unmineralized osteoid weakens bone structure, resulting in increased risk of deformity and fractures. Think of it like unbaked cookie dough versus a properly baked cookie – the dough lacks structural integrity.
- Diffuse Bone Pain and Tenderness: This is a common presenting complaint, but it's important to remember that it's non-specific. Many other conditions cause bone pain.
- Proximal Muscle Weakness: Weakness in the muscles of the shoulders and hips is another key symptom.
- Undiagnosed Cases: A significant number of cases likely go undetected due to the nonspecific nature of symptoms, emphasizing the need for thorough investigation when appropriate.
- Excessive Unmineralized Osteoid: Microscopic examination reveals bony trabeculae (bone struts) covered by a significantly thicker-than-normal layer of unmineralized osteoid. This is the definitive histopathological finding confirming the diagnosis.
- Treatment & Prognosis: Vitamin D supplementation typically leads to rapid bone mineralization and symptom resolution. However, some bone deformities may persist.
- Osteoid: The unmineralized organic matrix of bone. It's the "building material" that should harden with calcium and phosphate.
- Non-specific Symptoms: Osteomalacia's symptoms are not unique to the disease, making diagnosis challenging.
- Vitamin D's Crucial Role: Vitamin D is essential for calcium absorption and bone mineralization.

- Published on
Pathology - Osteoporosis:
.
I. Definition & Epidemiology:
.
I. Definition & Epidemiology:
- Definition: Osteoporosis is a metabolic bone disease characterized by decreased bone mass, increased bone fragility, and a heightened risk of fractures. It's a silent disease often undetected until a fracture occurs.
- Epidemiology: Extremely common, predominantly affecting elderly women, although it can occur in all ages.
- Primary Cause: Estrogen deficiency (particularly post-menopause in women) is a major risk factor.
- Other Contributing Factors:
- Glucocorticoid therapy (steroids)
- Cushing's syndrome
- Hyperparathyroidism
- Hyperthyroidism
- Celiac disease
- Inflammatory bowel disease
- Bone Mass Determination: Bone mass in later life is influenced by:
- Peak bone mass: Achieved during early adulthood (largely genetic, but modifiable by nutrition, physical activity, and early life health).
- Rate of bone loss: Increases with age due to:
- Decreased bone turnover
- Reduced physical activity
- Diminished calcium absorption (gut)
- Accelerated bone loss in women post-menopause (due to estrogen deficiency).
- Glucocorticoid Effect: Glucocorticoids negatively impact bone health by:
- Decreasing osteoblast activity and lifespan
- Reducing gut calcium absorption
- Increasing renal calcium loss
- Suppressing sex hormone production (further increasing bone loss).
- Often Asymptomatic: Most cases remain undiagnosed until a fracture occurs.
- Fracture Sites: Common fracture locations include:
- Vertebrae (leading to height loss and kyphosis – curvature of the spine) – can occur spontaneously or with minimal trauma.
- Distal radius (Colles' fracture) – typically from a fall.
- Neck of femur (hip fracture) – often from a fall from standing height or less.
- Cancellous Bone (spongy bone): Thinning and disconnection of trabeculae (supporting bone structures).
- Cortical Bone (compact bone): Thinning with enlargement of Haversian canals (channels containing blood vessels).
- Hip Fractures (Neck of Femur): Pose the most significant risk, often requiring hospitalization and surgery. Mortality risk is increased in elderly patients with other health problems.

- Published on
Pathology-Paget's Disease
I. Definition & Epidemiology:
I. Definition & Epidemiology:
- What is it? A metabolic bone disease characterized by excessive, disorganized bone turnover in specific skeletal areas. This means bone is broken down and rebuilt at an abnormally high rate, resulting in weakened, deformed bone.
- Who gets it? Primarily older adults. Geographic distribution is uneven, with higher prevalence in the UK.
- Cause: Unknown. A viral etiology is suspected based on viral inclusions observed in osteoclasts (bone-resorbing cells) under electron microscopy, but this isn't definitively proven.
- How it develops (Pathogenesis): The disease progresses through stages (which may overlap):
- Increased Osteoclastic Resorption: Excessive breakdown of bone by osteoclasts.
- Increased Osteoblastic Formation: Frantic, but disorganized, bone formation by osteoblasts attempting to compensate.
- Abnormal Bone Formation: The bone formed is thick but weak, leading to deformities and fractures.
- Most Patients (Asymptomatic): Often diagnosed incidentally through radiological findings (X-rays, etc.).
- Symptomatic Patients: Typically present with bone pain and deformity.
- Elevated Serum Alkaline Phosphatase: A key indicator due to the intense osteoblastic activity. This enzyme is involved in bone formation.
- Normal Serum Calcium: Usually, blood calcium levels remain within the normal range.
- Thickened Trabeculae: Bone beams are abnormally thick, with a "jigsaw" pattern of cement lines (indicating repeated cycles of bone resorption and formation).
- Altered Haversian Canals: Irregular trabeculae replace the normal Haversian canals (channels within the bone).
- Fibrotic Marrow: The bone marrow becomes densely fibrous.
- Generally Favorable: Most individuals experience no significant problems.
- Potential Complications:
- Pathological Fractures: Fractures due to weakened bone.
- Deafness: Enlarged skull bones can compress cranial nerve VIII (vestibulocochlear nerve), leading to hearing loss.
- Osteosarcoma: This is a serious bone cancer. Though rare (<1% of cases), it's associated with a poor prognosis and should be considered if patient paget's disease experiences rapidly worsening bone pain.< />pan>

