- Published on
Diagnostic Tests: Serum Protein Electrophoresis (SPEP)
Overview — What SPEP Is?
Serum protein electrophoresis (SPEP) is a laboratory test that separates and quantifies the major protein components of the blood. By applying an electric current to serum, proteins migrate based on their size and charge, producing a pattern that distinguishes:
- Albumin
- α1, α2, β, and γ globulin fractions
- Immunoglobulins (IgG, IgA, IgM, etc.)
This allows clinicians to identify abnormal elevations—especially monoclonal spikes (“M-spikes”) that suggest plasma cell disorders.
When SPEP Is the Correct Answer
Choose SPEP when evaluating:
- Elevated total serum protein
- Unexplained anemia, especially with rouleaux formation
- Bone pain or lytic lesions on X-ray
- Renal failure of unclear origin
- Symptoms suggestive of plasma cell dyscrasias (fatigue, weight loss, recurrent infections)
SPEP is the best initial test to diagnose multiple myeloma, particularly when:
- X-rays for lytic lesions have already been performed
- Or when imaging is not an available answer choice
Most Common Reason for an Abnormal SPEP
The most common cause of an abnormal SPEP—particularly a monoclonal spike—is:
Monoclonal Gammopathy of Unknown/Undetermined Significance (MGUS)
MGUS is far more common than multiple myeloma and is often asymptomatic, although it requires monitoring due to its risk of progression.
Most Accurate Diagnostic Test
For confirming an IgG monoclonal abnormality detected on SPEP, the most accurate test is:
Bone Marrow Biopsy
Findings diagnostic of multiple myeloma include:
- >10% clonal plasma cells
- Evidence of organ damage (CRAB criteria: hyperCalcemia, Renal failure, Anemia, Bone lesions)
If you’d like, I can produce a quick comparison table: SPEP vs. UPEP vs. Immunofixation, or a high-yield myeloma diagnostic algorithm.

- Published on
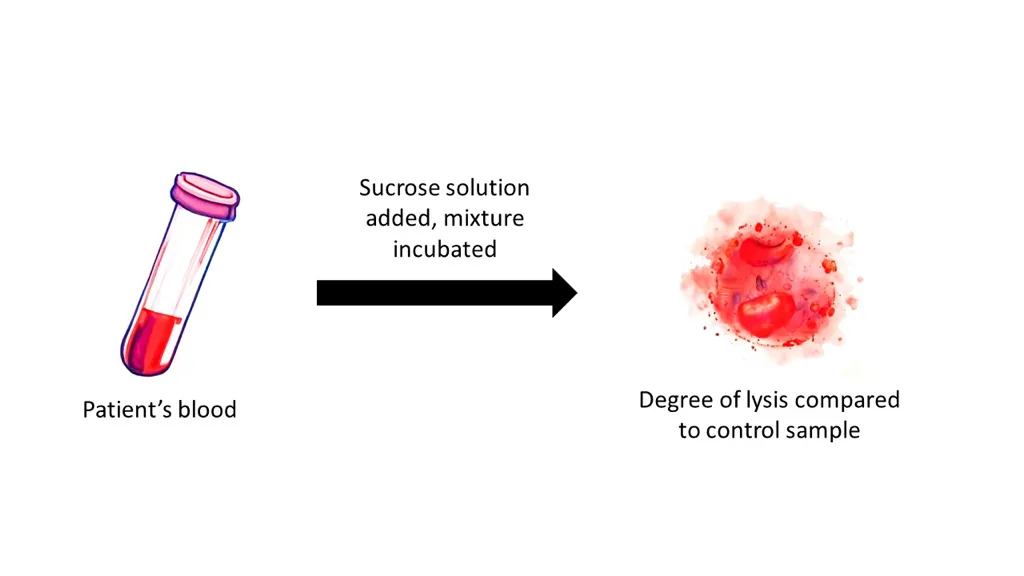
Diagnostic Tests: Sucrose Lysis Test
Overview — What This Test Is
The sucrose lysis test is a screening test for paroxysmal nocturnal hemoglobinuria (PNH).
In this test, the patient’s red blood cells are placed in a sucrose-containing solution, which promotes complement activation. PNH RBCs are unusually sensitive to complement-mediated destruction, so the presence of hemolysis in this solution suggests PNH.
The mechanism:
When to Answer “Sucrose Lysis Test”
Choose the sucrose lysis test in a clinical scenario that describes the classic features of PNH, such as:
When these features appear together, the sucrose lysis test is the classic screening test.
Most Accurate Diagnostic Test
The most accurate and modern diagnostic test for PNH is:
Flow Cytometry for CD55 and CD59 Deficiency
Flow cytometry is the gold standard because it directly demonstrates the absence of these protective markers on RBCs and WBCs.
Overview — What This Test Is
The sucrose lysis test is a screening test for paroxysmal nocturnal hemoglobinuria (PNH).
In this test, the patient’s red blood cells are placed in a sucrose-containing solution, which promotes complement activation. PNH RBCs are unusually sensitive to complement-mediated destruction, so the presence of hemolysis in this solution suggests PNH.
The mechanism:
- PNH cells lack protective surface proteins (CD55, CD59) that normally prevent complement-mediated lysis.
- Sucrose enhances complement binding → RBC destruction → hemolysis detected.
When to Answer “Sucrose Lysis Test”
Choose the sucrose lysis test in a clinical scenario that describes the classic features of PNH, such as:
- Dark urine in the morning (from overnight hemoglobinuria)
- Episodic hemolysis
- Pancytopenia
- Venous thrombosis, especially in unusual sites such as hepatic, portal, or cerebral veins
- Negative Coombs test (helps distinguish PNH from autoimmune hemolysis)
When these features appear together, the sucrose lysis test is the classic screening test.
Most Accurate Diagnostic Test
The most accurate and modern diagnostic test for PNH is:
Flow Cytometry for CD55 and CD59 Deficiency
- These proteins are also called decay-accelerating factor (DAF) and membrane inhibitor of reactive lysis (MIRL).
- PNH cells are missing CD55 and CD59 due to a mutation in the PIGA gene affecting the GPI anchor that normally attaches these protective proteins to the cell surface.
Flow cytometry is the gold standard because it directly demonstrates the absence of these protective markers on RBCs and WBCs.
- Published on
Diagnostic Tests-Tests: D-Dimer and Fibrin Split Products
Overview
D-dimer and fibrin split products (FSPs) are laboratory markers that reflect activation of the coagulation and fibrinolytic systems. D-dimer is generated when plasmin degrades cross-linked fibrin, whereas fibrin split products are formed when thrombin acts on fibrinogen, producing degradation fragments. Both markers become clinically significant when elevated, as they indicate abnormal or excessive clot formation and breakdown within the body.
Measurement Methods
D-dimer levels can be measured through two commonly used laboratory techniques:
Fibrin split products are measured through similar immunologic methods, but they are primarily ordered in the evaluation of disseminated intravascular coagulation (DIC).
What These Tests Detect
Elevated D-dimer and FSP levels indicate pathologic clot formation and subsequent fibrinolysis. When the coagulation cascade is activated, fibrin clots form and are later broken down, generating measurable fragments.
Importantly, these tests do not assess platelet function and provide no information about platelet aggregation or adhesion disorders.
Interpretation of Test Results
When to Order These Tests
D-dimer and fibrin split products should be ordered in the following scenarios:
Overview
D-dimer and fibrin split products (FSPs) are laboratory markers that reflect activation of the coagulation and fibrinolytic systems. D-dimer is generated when plasmin degrades cross-linked fibrin, whereas fibrin split products are formed when thrombin acts on fibrinogen, producing degradation fragments. Both markers become clinically significant when elevated, as they indicate abnormal or excessive clot formation and breakdown within the body.
Measurement Methods
D-dimer levels can be measured through two commonly used laboratory techniques:
- Latex agglutination assay:
A rapid test, useful for quick screening but less sensitive. - ELISA (enzyme-linked immunosorbent assay):
The most accurate and far more sensitive method, capable of detecting very low levels of D-dimer. Because of its high sensitivity, the ELISA is the preferred test when ruling out thrombotic conditions such as deep vein thrombosis (DVT) or pulmonary embolism (PE).
Fibrin split products are measured through similar immunologic methods, but they are primarily ordered in the evaluation of disseminated intravascular coagulation (DIC).
What These Tests Detect
Elevated D-dimer and FSP levels indicate pathologic clot formation and subsequent fibrinolysis. When the coagulation cascade is activated, fibrin clots form and are later broken down, generating measurable fragments.
Importantly, these tests do not assess platelet function and provide no information about platelet aggregation or adhesion disorders.
Interpretation of Test Results
- Elevated levels suggest active clot formation and breakdown and are characteristic of conditions such as:
- Disseminated intravascular coagulation (DIC)
- Recent surgery or trauma
- Infection or sepsis
- Malignancy
- Venous thromboembolism (DVT or PE)
- A negative ELISA D-dimer is powerful for ruling out DVT or PE in patients with a low pretest probability.
- However, a positive D-dimer is not diagnostic of DVT or PE because many conditions can elevate it.
- In DIC, both D-dimer and fibrin split products are elevated, reflecting uncontrolled coagulation and fibrinolysis.
When to Order These Tests
D-dimer and fibrin split products should be ordered in the following scenarios:
- To evaluate suspected DIC, particularly when accompanied by thrombocytopenia, prolonged PT/aPTT, and clinical evidence of bleeding or thrombosis.
- To rule out pulmonary embolism or deep vein thrombosis, but only in patients with low pretest probability, where a negative ELISA D-dimer effectively excludes the diagnosis.
- As supportive evidence in conditions involving widespread clot formation and fibrinolysis.
- Published on
Diagnostics Tests: Hypersegmented Neutrophils
Overview
A hypersegmented neutrophil is a neutrophil with an abnormally high number of nuclear lobes. Normal neutrophils average about 3–4 lobes, but when the average exceeds 4 lobes, or when 5% of cells have ≥5 lobes, or when even a single 6-lobed neutrophil is present, the finding is considered hypersegmentation. This morphological change is a classic hematologic clue and serves as a defining feature of megaloblastic anemia.
Associated Diseases
Hypersegmented neutrophils are most commonly seen in:
These deficiencies impair DNA synthesis, leading to delayed nuclear maturation in hematopoietic cells. The result is macrocytosis, ineffective erythropoiesis, and the hallmark hypersegmented neutrophils seen on peripheral blood smear. These findings may accompany clinical features like glossitis, anemia, and—in B12 deficiency—neurologic symptoms.
Most Accurate Diagnostic Tests
To confirm megaloblastic anemia and determine its cause, the following tests are most accurate:
Therapy
Treatment depends on the underlying deficiency:
Overview
A hypersegmented neutrophil is a neutrophil with an abnormally high number of nuclear lobes. Normal neutrophils average about 3–4 lobes, but when the average exceeds 4 lobes, or when 5% of cells have ≥5 lobes, or when even a single 6-lobed neutrophil is present, the finding is considered hypersegmentation. This morphological change is a classic hematologic clue and serves as a defining feature of megaloblastic anemia.
Associated Diseases
Hypersegmented neutrophils are most commonly seen in:
- Vitamin B12 deficiency
- Folate deficiency
These deficiencies impair DNA synthesis, leading to delayed nuclear maturation in hematopoietic cells. The result is macrocytosis, ineffective erythropoiesis, and the hallmark hypersegmented neutrophils seen on peripheral blood smear. These findings may accompany clinical features like glossitis, anemia, and—in B12 deficiency—neurologic symptoms.
Most Accurate Diagnostic Tests
To confirm megaloblastic anemia and determine its cause, the following tests are most accurate:
- Serum vitamin B12 and folate levels – first-line and most direct assessment.
- Methylmalonic acid (MMA) – elevated in B12 deficiency only, not folate deficiency, making it helpful for distinguishing between the two.
- Homocysteine levels – elevated in both B12 and folate deficiency (helps support the diagnosis, but not specific).
- Anti–intrinsic factor antibodies and antiparietal cell antibodies – used to identify pernicious anemia, the classic autoimmune cause of B12 deficiency.
Therapy
Treatment depends on the underlying deficiency:
- Vitamin B12 deficiency:
- Replacement with intramuscular B12 injections for severe deficiency or malabsorption
- Oral B12 for mild cases or dietary deficiency
- Lifelong therapy is required for pernicious anemia
- Folate deficiency:
- Oral folic acid supplementation
- Dietary improvement
- Important note: Never treat with folate alone if B12 deficiency is possible, as folate can correct the anemia while allowing neurologic damage from B12 deficiency to persist or worsen.

- Published on
Diagnostic Tests: Hairy Cell Leukemia (Blood Smear Findings)
Overview — What the Blood Smear Shows
The blood smear displays lymphoid cells with characteristic cytoplasmic projections, known as “hairy cells.” These projections give the cells a fuzzy or “hairy” appearance, which is the classic morphologic hallmark of hairy cell leukemia (HCL). This is a rare, chronic B-cell lymphoproliferative disorder.
Clinical Scenario — When to Suspect and Answer Hairy Cell Leukemia
Hairy cell leukemia should be strongly suspected when the clinical presentation includes:
This combination—middle-aged patient, massive splenomegaly, pancytopenia, and characteristic smear findings—points to hairy cell leukemia.
Most Accurate Diagnostic Test
The most accurate test for confirming hairy cell leukemia is:
Tartrate-Resistant Acid Phosphatase (TRAP) positivity
(Modern practice also uses flow cytometry markers such as CD11c, CD25, CD103, and detection of BRAF V600E mutation, but TRAP is the classic board-test answer.)
If you’d like, I can also create a rapid diagnostic table comparing hairy cell leukemia with CLL, AML, and other pancytopenia causes.
Clinical Scenario — When to Suspect and Answer Hairy Cell Leukemia
Hairy cell leukemia should be strongly suspected when the clinical presentation includes:
This combination—middle-aged patient, massive splenomegaly, pancytopenia, and characteristic smear findings—points to hairy cell leukemia.
Overview — What the Blood Smear Shows
The blood smear displays lymphoid cells with characteristic cytoplasmic projections, known as “hairy cells.” These projections give the cells a fuzzy or “hairy” appearance, which is the classic morphologic hallmark of hairy cell leukemia (HCL). This is a rare, chronic B-cell lymphoproliferative disorder.
Clinical Scenario — When to Suspect and Answer Hairy Cell Leukemia
Hairy cell leukemia should be strongly suspected when the clinical presentation includes:
- A middle-aged man (most common demographic)
- Gradual onset of fatigue, weakness, or recurrent infections
- Massive splenomegaly, often the dominant physical finding
- Hepatomegaly (present in about 50% of patients)
- Pancytopenia, which is the hallmark laboratory abnormality
- Particularly monocytopenia
- Bone marrow often becomes fibrotic and difficult to aspirate (“dry tap”)
This combination—middle-aged patient, massive splenomegaly, pancytopenia, and characteristic smear findings—points to hairy cell leukemia.
Most Accurate Diagnostic Test
The most accurate test for confirming hairy cell leukemia is:
Tartrate-Resistant Acid Phosphatase (TRAP) positivity
- Performed on a bone marrow biopsy
- Hairy cells show strong acid phosphatase activity that remains positive even in the presence of tartrate, distinguishing them from other leukemic cells.
(Modern practice also uses flow cytometry markers such as CD11c, CD25, CD103, and detection of BRAF V600E mutation, but TRAP is the classic board-test answer.)
If you’d like, I can also create a rapid diagnostic table comparing hairy cell leukemia with CLL, AML, and other pancytopenia causes.
Clinical Scenario — When to Suspect and Answer Hairy Cell Leukemia
Hairy cell leukemia should be strongly suspected when the clinical presentation includes:
- A middle-aged man (most common demographic)
- Gradual onset of fatigue, weakness, or recurrent infections
- Massive splenomegaly, often the dominant physical finding
- Hepatomegaly (present in about 50% of patients)
- Pancytopenia, which is the hallmark laboratory abnormality
- Particularly monocytopenia
- Bone marrow often becomes fibrotic and difficult to aspirate (“dry tap”)
This combination—middle-aged patient, massive splenomegaly, pancytopenia, and characteristic smear findings—points to hairy cell leukemia.

- Published on
Diagnostic Tests: Peripheral Smear Findings in G6PD Deficiency
Overview — What the Peripheral Smear Shows
This peripheral smear demonstrates Heinz bodies, which are inclusions formed by precipitated, oxidized hemoglobin inside red blood cells. These occur when RBCs are exposed to oxidative stress in individuals with glucose-6-phosphate dehydrogenase (G6PD) deficiency.
G6PD deficiency is an X-linked recessive condition and is particularly common in African-American males (10–15%).
Clinical Scenario — When This Is the Correct Answer
You should suspect G6PD deficiency when the presentation describes a previously healthy male who suddenly develops signs of acute intravascular hemolysis after oxidative stress. Key features include:
Common triggers of oxidative stress include:
When a question stem includes these triggers plus acute hemolysis and the smear shows characteristic RBC inclusions, G6PD deficiency is the diagnosis.
Other Cells Characteristic of G6PD Deficiency
Once Heinz bodies form, the spleen attempts to remove them. This “pitting” process produces:
Bite cells (degmacytes)
Definitive Diagnostic Test
The definitive test for confirming G6PD deficiency is:
Measurement of G6PD enzyme level
Important caveat:
You must wait approximately 2 months after the hemolytic episode to test.
Immediately after hemolysis, the older, most deficient red cells have already been destroyed. The remaining population consists of young reticulocytes with relatively higher G6PD activity, which can give a false-normal result.
If you’d like, I can also create a comparison chart of G6PD deficiency vs. other hemolytic anemias (like autoimmune hemolysis or sickle cell), or provide a rapid-review summary for exams.
Overview — What the Peripheral Smear Shows
This peripheral smear demonstrates Heinz bodies, which are inclusions formed by precipitated, oxidized hemoglobin inside red blood cells. These occur when RBCs are exposed to oxidative stress in individuals with glucose-6-phosphate dehydrogenase (G6PD) deficiency.
G6PD deficiency is an X-linked recessive condition and is particularly common in African-American males (10–15%).
Clinical Scenario — When This Is the Correct Answer
You should suspect G6PD deficiency when the presentation describes a previously healthy male who suddenly develops signs of acute intravascular hemolysis after oxidative stress. Key features include:
- Weakness and fatigue
- Tachycardia
- Jaundice
- Dark urine (hemoglobinuria)
- Sudden drop in hemoglobin
Common triggers of oxidative stress include:
- Infections (most common cause)
- Drugs, including:
- Sulfa drugs
- Primaquine
- Dapsone
- Quinidine
- Nitrofurantoin
- Fava beans (favism)
When a question stem includes these triggers plus acute hemolysis and the smear shows characteristic RBC inclusions, G6PD deficiency is the diagnosis.
Other Cells Characteristic of G6PD Deficiency
Once Heinz bodies form, the spleen attempts to remove them. This “pitting” process produces:
Bite cells (degmacytes)
- RBCs appear as though a bite has been taken out of them.
- These cells are classic for G6PD deficiency and often accompany Heinz body formation.
Definitive Diagnostic Test
The definitive test for confirming G6PD deficiency is:
Measurement of G6PD enzyme level
Important caveat:
You must wait approximately 2 months after the hemolytic episode to test.
Immediately after hemolysis, the older, most deficient red cells have already been destroyed. The remaining population consists of young reticulocytes with relatively higher G6PD activity, which can give a false-normal result.
If you’d like, I can also create a comparison chart of G6PD deficiency vs. other hemolytic anemias (like autoimmune hemolysis or sickle cell), or provide a rapid-review summary for exams.
- Published on
Diagnostic Tests: Fragmented Red Blood Cells (Schistocytes)
Overview — What This Is
The smear shows fragmented red blood cells, also referred to as schistocytes or helmet cells. These irregularly shaped RBC fragments form when erythrocytes are mechanically sheared within the circulation. Their presence is a hallmark of intravascular hemolysis and defines microangiopathic hemolytic anemia (MAHA).
Diseases Associated With Schistocytes
Fragmented RBCs occur in conditions where red cells are destroyed while passing through areas of fibrin deposition, turbulence, or mechanical injury. Major associated disorders include:
- Thrombotic Thrombocytopenic Purpura (TTP)
- Hemolytic Uremic Syndrome (HUS)
- Disseminated Intravascular Coagulation (DIC)
- Major blood group incompatibility (e.g., ABO mismatch)
- Paroxysmal Nocturnal Hemoglobinuria (PNH)
- Mechanical destruction from:
- Artificial heart valves
- Mechanical circulatory support devices (e.g., LVADs)
- Snake bites (due to hemotoxic venom effects)
These conditions share a common mechanism—red cell fragmentation within the vascular system.
Associated Laboratory Abnormalities
Because schistocytes signify intravascular hemolysis, several classic lab abnormalities accompany their presence:
- Elevated LDH — released from destroyed RBCs
- Elevated indirect (unconjugated) bilirubin — from heme breakdown
- Elevated reticulocyte count — bone marrow compensation
- Decreased haptoglobin — binds free hemoglobin, becomes depleted in intravascular hemolysis
- Published on
Diagnostic Tests: Auer Rods
Overview — What This Is
The structure shown is an Auer rod, which is an eosinophilic, needle-shaped cytoplasmic inclusion found within myeloblasts or promyelocytes. These inclusions are composed of abnormal azurophilic granules and represent crystalline aggregates of myeloperoxidase. Their presence indicates a malignant proliferation of myeloid precursors.
Disease Associated With Auer Rods
Auer rods are pathognomonic for acute myelogenous leukemia (AML).
They may appear in several AML subtypes, but are especially prominent in:
• Acute Promyelocytic Leukemia (APL, M3) — often showing bundles of Auer rods (“faggot cells”), which is a high-yield test clue.
Their presence confirms the myeloid origin of the malignant blasts
When This Is the Correct Answer
You should answer Auer rods / AML when the question presents a patient with:
• Pancytopenia (fatigue, infections, bleeding)
• Peripheral smear showing blasts
• >20% blasts on bone marrow biopsy, meeting diagnostic criteria for acute leukemia
• Systemic symptoms such as fatigue, fever, infections, bruising, or bleeding
Additional diagnostic support includes:
• Myeloperoxidase (MPO) positivity on histochemical staining
• Expression of myeloid markers: CD13, CD33, CD117
• Presence of Auer rods confirms AML over ALL or other hematologic malignancies
In APL (t(15;17)), Auer rods are particularly dense and clinically important because the disease is associated with DIC, and treatment requires all-trans retinoic acid (ATRA).
Overview — What This Is
The structure shown is an Auer rod, which is an eosinophilic, needle-shaped cytoplasmic inclusion found within myeloblasts or promyelocytes. These inclusions are composed of abnormal azurophilic granules and represent crystalline aggregates of myeloperoxidase. Their presence indicates a malignant proliferation of myeloid precursors.
Disease Associated With Auer Rods
Auer rods are pathognomonic for acute myelogenous leukemia (AML).
They may appear in several AML subtypes, but are especially prominent in:
• Acute Promyelocytic Leukemia (APL, M3) — often showing bundles of Auer rods (“faggot cells”), which is a high-yield test clue.
Their presence confirms the myeloid origin of the malignant blasts
When This Is the Correct Answer
You should answer Auer rods / AML when the question presents a patient with:
• Pancytopenia (fatigue, infections, bleeding)
• Peripheral smear showing blasts
• >20% blasts on bone marrow biopsy, meeting diagnostic criteria for acute leukemia
• Systemic symptoms such as fatigue, fever, infections, bruising, or bleeding
Additional diagnostic support includes:
• Myeloperoxidase (MPO) positivity on histochemical staining
• Expression of myeloid markers: CD13, CD33, CD117
• Presence of Auer rods confirms AML over ALL or other hematologic malignancies
In APL (t(15;17)), Auer rods are particularly dense and clinically important because the disease is associated with DIC, and treatment requires all-trans retinoic acid (ATRA).
- Published on
Diagnostic Tests: Ringed Sideroblasts
Overview — What This Is
The cell shown is a ringed sideroblast, a pathological erythroblast with iron-loaded mitochondria encircling the nucleus. These iron deposits can only be seen clearly using a Prussian blue stain, which highlights the mitochondrial iron in a ring-like pattern. Ringed sideroblasts reflect a failure of the developing red blood cell to properly incorporate iron into heme during erythropoiesis.
What Causes This?
Ringed sideroblasts form when iron accumulates in mitochondria because of defective heme synthesis within erythroid precursors. This impairment leads to:
Common causes of acquired sideroblastic anemia include:
Associated Hematologic Disorders
Ringed sideroblasts are the defining feature of sideroblastic anemia, but they are also associated with other disorders, including:
Thus, their presence should prompt evaluation for toxin exposure, alcohol use, medications, and underlying marrow disorders such as MDS.
If you’d like, I can create a comparison of sideroblastic anemia vs. iron deficiency anemia or provide a quick diagnostic algorithm for microcytic anemias.
Overview — What This Is
The cell shown is a ringed sideroblast, a pathological erythroblast with iron-loaded mitochondria encircling the nucleus. These iron deposits can only be seen clearly using a Prussian blue stain, which highlights the mitochondrial iron in a ring-like pattern. Ringed sideroblasts reflect a failure of the developing red blood cell to properly incorporate iron into heme during erythropoiesis.
What Causes This?
Ringed sideroblasts form when iron accumulates in mitochondria because of defective heme synthesis within erythroid precursors. This impairment leads to:
- Microcytic anemia
- Elevated serum iron level
- Ineffective erythropoiesis
Common causes of acquired sideroblastic anemia include:
- Alcohol use (most common)
- Drugs, such as:
- Isoniazid (INH)
- Chloramphenicol
- Toxins, including:
- Lead
- Zinc
Associated Hematologic Disorders
Ringed sideroblasts are the defining feature of sideroblastic anemia, but they are also associated with other disorders, including:
- Myelodysplastic Syndrome (MDS) — particularly the subtype known as refractory anemia with ring sideroblasts (RARS)
- Congenital sideroblastic anemias (less common, involve mutations in heme synthesis enzymes)
Thus, their presence should prompt evaluation for toxin exposure, alcohol use, medications, and underlying marrow disorders such as MDS.
If you’d like, I can create a comparison of sideroblastic anemia vs. iron deficiency anemia or provide a quick diagnostic algorithm for microcytic anemias.
- Published on
Diagnostic Tests: Target Cells
Overview — What These Are?
The cells shown are target cells (codocytes). Target cells have a characteristic “bull’s-eye” appearance caused by an excess of cell membrane relative to cell volume. This altered surface-area–to–volume ratio creates a central area of hemoglobin surrounded by a pale ring and then an outer ring of hemoglobin.
Most Accurate Diagnostic Test
The most accurate diagnostic test when evaluating conditions associated with target cells is:
Hemoglobin Electrophoresis
This test helps identify hemoglobinopathies such as:
- Sickle cell disease
- Hemoglobin C disease
- Thalassemia
Electrophoresis will distinguish between different abnormal hemoglobins and determine specific genetic variants.
Associated Diseases
Target cells may appear in several hematologic and systemic conditions, including:
- Hemoglobinopathies
- Hemoglobin C disease (most strongly associated)
- Sickle cell disease (HbSS)
- Thalassemias (α and β)
- Liver disease
Due to increased membrane lipids, resulting in excess surface area. - Iron deficiency anemia
Though less classic, target cells may be seen in severe cases.
Do Target Cells Have a Shorter Survival?
Despite their abnormal morphology, target cells do NOT have a shorter survival than normal red blood cells.
They are structurally abnormal but not inherently more fragile, unlike spherocytes or sickled cells.

