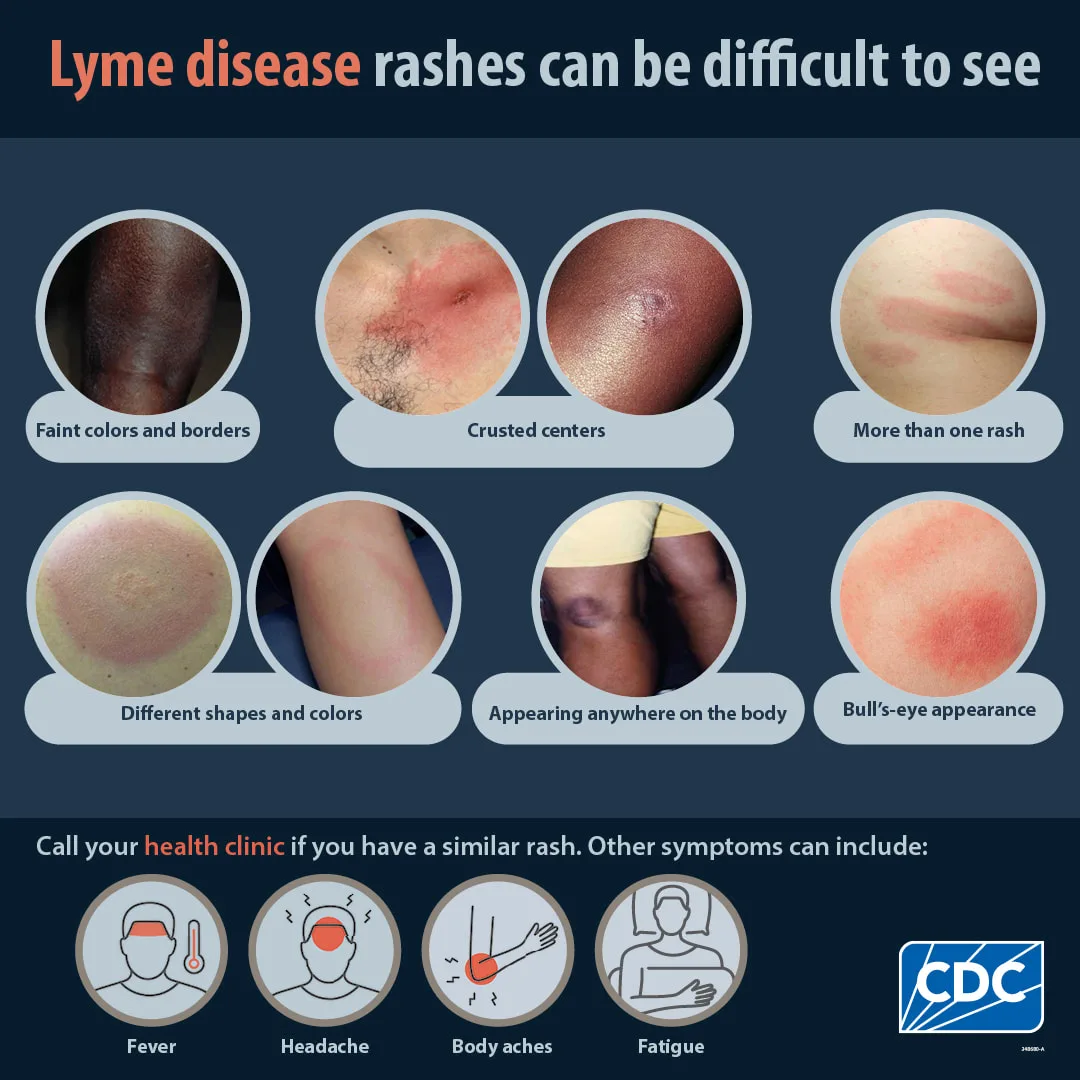
- Published on
KembaraXtra-Medicine – Intestinal Malrotation
Intestinal malrotation is a congenital condition resulting from incomplete rotation and fixation of the intestine during embryogenesis, specifically during the transition from the extracolonic position around the tenth week of gestation. It is commonly associated with heterotaxia syndromes and frequently occurs alongside other congenital anomalies. Gastrointestinal associations include duodenal stenosis or atresia, duodenal web, Meckel diverticulum, intussusception, gastroesophageal reflux, omphalocele, gastroschisis, congenital diaphragmatic hernia, abdominal wall defects, and Hirschsprung disease. Congenital cardiac anomalies are present in approximately 27% of patients and significantly increase morbidity.
The underlying pathology involves abnormal positioning of the duodenojejunal junction, which remains to the right of the midline, and malposition of the cecum, which is often located in the upper left abdomen with abnormal mesenteric attachments. This abnormal anatomy predisposes patients to bowel obstruction and volvulus. Midgut volvulus is a serious complication that occurs when the small intestine twists around the superior mesenteric artery and vein, leading to vascular compromise and potential bowel ischemia. Malrotation is found in combination with other congenital anomalies in up to 70% of cases, particularly involving the cardiac, esophageal, urinary, and anorectal systems.
Malrotation occurs in approximately 1 in 500 live births. It is more common in males, with a male-to-female ratio of 2:1 in neonates. About 75% of cases are diagnosed during the newborn period, and 90% are identified by one year of age, although presentation can occur in adulthood. Mortality in infants can be as high as 24%, and the presence of necrotic bowel at surgery increases mortality risk significantly.
Clinical presentation varies by age. Neonates typically present with bilious vomiting, abdominal distention, bloody stools, constipation or obstipation, feeding difficulties, and poor weight gain. Infants and children older than one year may present with abdominal pain followed by bilious emesis. Older children and adolescents often experience chronic vomiting, intermittent colicky abdominal pain, diarrhea, hematemesis, or constipation. Adults usually present with vague and nonspecific gastrointestinal symptoms. Notably, up to 75% of patients may have a normal physical examination at presentation. Severe cases may show signs of dehydration, metabolic acidosis, peritonitis, ischemic bowel, sepsis, or shock.
Diagnosis is suggested by clinical history and physical examination and is confirmed through imaging, most commonly contrast radiography. Laboratory evaluation includes complete blood count, venous blood gas, electrolytes, renal function tests, glucose, coagulation profile, lactate, and type and screen. Plain abdominal radiographs are diagnostic in fewer than 30% of cases but may show signs suggestive of volvulus, such as duodenal obstruction, gastric distention, paucity of distal bowel gas, generalized small-bowel distention, or a double-bubble sign. Upper gastrointestinal contrast studies are the diagnostic test of choice, with high sensitivity and accuracy, demonstrating abnormal position of the duodenojejunal junction, corkscrew appearance of the duodenum in volvulus, or right-sided jejunum. Contrast enema may help identify cecal position but has a significant false-negative rate. Ultrasound can demonstrate abnormal superior mesenteric artery–vein relationships and the classic whirlpool sign in volvulus, although a normal study does not exclude malrotation. CT imaging is generally reserved for adults.
Differential diagnosis depends on age and presentation. In early life, considerations include Hirschsprung disease, necrotizing enterocolitis, and intussusception. In children with acute abdominal pain and peritoneal signs, appendicitis and sepsis must be considered. Older children and adults with chronic or vague symptoms may be misdiagnosed with irritable bowel syndrome, peptic ulcer disease, biliary or pancreatic disorders, or psychiatric conditions.
Management of malrotation, particularly with midgut volvulus, is a surgical emergency. Initial stabilization focuses on airway, breathing, and circulation, with rapid intravenous fluid resuscitation to correct hypovolemia and metabolic acidosis. Broad-spectrum antibiotics are initiated if there are signs of sepsis or peritonitis. Nasogastric decompression and prompt surgical consultation are essential. Definitive treatment involves emergent surgical correction, including detorsion of the volvulus, restoration of intestinal perfusion, resection of necrotic bowel if present, and reassessment of bowel viability when necessary. Patients are kept nil per os and require close postoperative monitoring.
Hospital admission is indicated for acute abdomen, need for surgical intervention, significant dehydration, acidosis, sepsis, or shock. Discharge is uncommon and reserved for stable, asymptomatic patients with incidental findings after thorough pediatric surgical evaluation. Early recognition, rapid resuscitation, and timely referral to a facility with pediatric surgical expertise are critical to improving outcomes and reducing morbidity and mortality.
Intestinal malrotation is a congenital condition resulting from incomplete rotation and fixation of the intestine during embryogenesis, specifically during the transition from the extracolonic position around the tenth week of gestation. It is commonly associated with heterotaxia syndromes and frequently occurs alongside other congenital anomalies. Gastrointestinal associations include duodenal stenosis or atresia, duodenal web, Meckel diverticulum, intussusception, gastroesophageal reflux, omphalocele, gastroschisis, congenital diaphragmatic hernia, abdominal wall defects, and Hirschsprung disease. Congenital cardiac anomalies are present in approximately 27% of patients and significantly increase morbidity.
The underlying pathology involves abnormal positioning of the duodenojejunal junction, which remains to the right of the midline, and malposition of the cecum, which is often located in the upper left abdomen with abnormal mesenteric attachments. This abnormal anatomy predisposes patients to bowel obstruction and volvulus. Midgut volvulus is a serious complication that occurs when the small intestine twists around the superior mesenteric artery and vein, leading to vascular compromise and potential bowel ischemia. Malrotation is found in combination with other congenital anomalies in up to 70% of cases, particularly involving the cardiac, esophageal, urinary, and anorectal systems.
Malrotation occurs in approximately 1 in 500 live births. It is more common in males, with a male-to-female ratio of 2:1 in neonates. About 75% of cases are diagnosed during the newborn period, and 90% are identified by one year of age, although presentation can occur in adulthood. Mortality in infants can be as high as 24%, and the presence of necrotic bowel at surgery increases mortality risk significantly.
Clinical presentation varies by age. Neonates typically present with bilious vomiting, abdominal distention, bloody stools, constipation or obstipation, feeding difficulties, and poor weight gain. Infants and children older than one year may present with abdominal pain followed by bilious emesis. Older children and adolescents often experience chronic vomiting, intermittent colicky abdominal pain, diarrhea, hematemesis, or constipation. Adults usually present with vague and nonspecific gastrointestinal symptoms. Notably, up to 75% of patients may have a normal physical examination at presentation. Severe cases may show signs of dehydration, metabolic acidosis, peritonitis, ischemic bowel, sepsis, or shock.
Diagnosis is suggested by clinical history and physical examination and is confirmed through imaging, most commonly contrast radiography. Laboratory evaluation includes complete blood count, venous blood gas, electrolytes, renal function tests, glucose, coagulation profile, lactate, and type and screen. Plain abdominal radiographs are diagnostic in fewer than 30% of cases but may show signs suggestive of volvulus, such as duodenal obstruction, gastric distention, paucity of distal bowel gas, generalized small-bowel distention, or a double-bubble sign. Upper gastrointestinal contrast studies are the diagnostic test of choice, with high sensitivity and accuracy, demonstrating abnormal position of the duodenojejunal junction, corkscrew appearance of the duodenum in volvulus, or right-sided jejunum. Contrast enema may help identify cecal position but has a significant false-negative rate. Ultrasound can demonstrate abnormal superior mesenteric artery–vein relationships and the classic whirlpool sign in volvulus, although a normal study does not exclude malrotation. CT imaging is generally reserved for adults.
Differential diagnosis depends on age and presentation. In early life, considerations include Hirschsprung disease, necrotizing enterocolitis, and intussusception. In children with acute abdominal pain and peritoneal signs, appendicitis and sepsis must be considered. Older children and adults with chronic or vague symptoms may be misdiagnosed with irritable bowel syndrome, peptic ulcer disease, biliary or pancreatic disorders, or psychiatric conditions.
Management of malrotation, particularly with midgut volvulus, is a surgical emergency. Initial stabilization focuses on airway, breathing, and circulation, with rapid intravenous fluid resuscitation to correct hypovolemia and metabolic acidosis. Broad-spectrum antibiotics are initiated if there are signs of sepsis or peritonitis. Nasogastric decompression and prompt surgical consultation are essential. Definitive treatment involves emergent surgical correction, including detorsion of the volvulus, restoration of intestinal perfusion, resection of necrotic bowel if present, and reassessment of bowel viability when necessary. Patients are kept nil per os and require close postoperative monitoring.
Hospital admission is indicated for acute abdomen, need for surgical intervention, significant dehydration, acidosis, sepsis, or shock. Discharge is uncommon and reserved for stable, asymptomatic patients with incidental findings after thorough pediatric surgical evaluation. Early recognition, rapid resuscitation, and timely referral to a facility with pediatric surgical expertise are critical to improving outcomes and reducing morbidity and mortality.
- Published on
Emergency and Acute Medicine – Lymphangitis
Basics description
Lymphangitis is an infection of the lymphatic vessels that drain an area of inflammation or infection. Histologically, lymphatic channels become dilated and filled with lymphocytes and histiocytes. Inflammation often extends into surrounding perilymphatic tissues and may progress to cellulitis or abscess formation if untreated.
Etiology
Acute lymphangitis is most commonly caused by bacterial infection, particularly group A β-hemolytic Streptococcus. Less common causes include other streptococcal species and Staphylococcus aureus, including community-associated methicillin-resistant S. aureus (CA-MRSA). CA-MRSA risk factors include prior MRSA infection, close household contact, military service, incarceration, contact sports, injection drug use, and men who have sex with men. CA-MRSA is now prevalent enough to warrant empiric coverage in many settings, especially in unresponsive or recurrent infections. Other causes include Pasteurella multocida from animal bites, Spirillum minus in rat-bite fever, and Wuchereria bancrofti in filariasis, particularly in immigrants from endemic regions. Chronic lymphangitis is usually due to fungal, mycobacterial, or parasitic infections, most commonly Sporothrix schenckii, acquired through gardening or farming injuries, and Mycobacterium marinum, associated with fish tanks and swimming pools.
Diagnosis signs and symptoms
Acute lymphangitis presents with warm, tender, erythematous streaks extending proximally from a primary site of infection toward regional lymph nodes. Associated findings include painful lymphadenopathy, peripheral edema of the involved extremity, and systemic symptoms such as fever, rigors, tachycardia, and headache. Chronic nodular lymphangitis typically presents with painless erythematous nodules, chancriform ulcers, or wart-like lesions at the inoculation site, with possible progression along lymphatic channels and minimal systemic symptoms.
Essential workup
Lymphangitis is primarily a clinical diagnosis based on history and physical examination, with attention directed toward identifying the source of infection.
Diagnosis tests and interpretation
Laboratory testing is often unnecessary, though leukocytosis may be present. Gram stain and culture of aspirate or biopsy from the most inflamed area are recommended in cases of treatment failure or when resistant organisms such as MRSA are suspected. Culture confirmation is essential when sporotrichosis or M. marinum infection is considered. Blood cultures may be positive in severe cases. Imaging is rarely required but plain radiographs may help identify abscesses, subcutaneous gas, or foreign bodies, and Doppler ultrasound may be useful to exclude deep venous thrombosis.
Differential diagnosis
The differential diagnosis includes superficial or deep thrombophlebitis, which typically lacks an initial infectious focus or regional lymphadenopathy, IV line infiltration, vaccine-related reactions, and phytophotodermatitis, which can cause linear inflammatory skin changes mimicking lymphangitis.
Treatment
Initial management focuses on stabilizing septic patients with airway support and resuscitation as needed. Antimicrobial therapy should be initiated promptly, with the first dose given in the emergency department. Treatment typically lasts 7–10 days and includes limb elevation, moist heat, and antibiotics guided by local resistance patterns. Empiric outpatient therapy often includes oral cephalexin plus trimethoprim–sulfamethoxazole to cover CA-MRSA, with alternatives such as clindamycin or doxycycline. Inpatient therapy may require IV nafcillin or equivalent agents. Animal bite–associated lymphangitis is treated with IV ampicillin–sulbactam. Chronic infections require organism-specific therapy, such as itraconazole or potassium iodide for sporotrichosis and combination antimycobacterial therapy for M. marinum.
Disposition and follow-up
Admission is indicated for toxic-appearing patients, those with immunosuppression, significant comorbidities, inability to tolerate oral therapy, or unreliable follow-up. Patients with mild disease who are clinically stable and able to take oral antibiotics may be discharged with close follow-up within 24–48 hours. Marking the borders of erythema before discharge helps assess treatment response.
Pearls and pitfalls
Empiric antibiotic therapy should include coverage for CA-MRSA in addition to streptococcal species. Failure to recognize resistant organisms or chronic infectious causes can lead to treatment failure and progression of disease.
Basics description
Lymphangitis is an infection of the lymphatic vessels that drain an area of inflammation or infection. Histologically, lymphatic channels become dilated and filled with lymphocytes and histiocytes. Inflammation often extends into surrounding perilymphatic tissues and may progress to cellulitis or abscess formation if untreated.
Etiology
Acute lymphangitis is most commonly caused by bacterial infection, particularly group A β-hemolytic Streptococcus. Less common causes include other streptococcal species and Staphylococcus aureus, including community-associated methicillin-resistant S. aureus (CA-MRSA). CA-MRSA risk factors include prior MRSA infection, close household contact, military service, incarceration, contact sports, injection drug use, and men who have sex with men. CA-MRSA is now prevalent enough to warrant empiric coverage in many settings, especially in unresponsive or recurrent infections. Other causes include Pasteurella multocida from animal bites, Spirillum minus in rat-bite fever, and Wuchereria bancrofti in filariasis, particularly in immigrants from endemic regions. Chronic lymphangitis is usually due to fungal, mycobacterial, or parasitic infections, most commonly Sporothrix schenckii, acquired through gardening or farming injuries, and Mycobacterium marinum, associated with fish tanks and swimming pools.
Diagnosis signs and symptoms
Acute lymphangitis presents with warm, tender, erythematous streaks extending proximally from a primary site of infection toward regional lymph nodes. Associated findings include painful lymphadenopathy, peripheral edema of the involved extremity, and systemic symptoms such as fever, rigors, tachycardia, and headache. Chronic nodular lymphangitis typically presents with painless erythematous nodules, chancriform ulcers, or wart-like lesions at the inoculation site, with possible progression along lymphatic channels and minimal systemic symptoms.
Essential workup
Lymphangitis is primarily a clinical diagnosis based on history and physical examination, with attention directed toward identifying the source of infection.
Diagnosis tests and interpretation
Laboratory testing is often unnecessary, though leukocytosis may be present. Gram stain and culture of aspirate or biopsy from the most inflamed area are recommended in cases of treatment failure or when resistant organisms such as MRSA are suspected. Culture confirmation is essential when sporotrichosis or M. marinum infection is considered. Blood cultures may be positive in severe cases. Imaging is rarely required but plain radiographs may help identify abscesses, subcutaneous gas, or foreign bodies, and Doppler ultrasound may be useful to exclude deep venous thrombosis.
Differential diagnosis
The differential diagnosis includes superficial or deep thrombophlebitis, which typically lacks an initial infectious focus or regional lymphadenopathy, IV line infiltration, vaccine-related reactions, and phytophotodermatitis, which can cause linear inflammatory skin changes mimicking lymphangitis.
Treatment
Initial management focuses on stabilizing septic patients with airway support and resuscitation as needed. Antimicrobial therapy should be initiated promptly, with the first dose given in the emergency department. Treatment typically lasts 7–10 days and includes limb elevation, moist heat, and antibiotics guided by local resistance patterns. Empiric outpatient therapy often includes oral cephalexin plus trimethoprim–sulfamethoxazole to cover CA-MRSA, with alternatives such as clindamycin or doxycycline. Inpatient therapy may require IV nafcillin or equivalent agents. Animal bite–associated lymphangitis is treated with IV ampicillin–sulbactam. Chronic infections require organism-specific therapy, such as itraconazole or potassium iodide for sporotrichosis and combination antimycobacterial therapy for M. marinum.
Disposition and follow-up
Admission is indicated for toxic-appearing patients, those with immunosuppression, significant comorbidities, inability to tolerate oral therapy, or unreliable follow-up. Patients with mild disease who are clinically stable and able to take oral antibiotics may be discharged with close follow-up within 24–48 hours. Marking the borders of erythema before discharge helps assess treatment response.
Pearls and pitfalls
Empiric antibiotic therapy should include coverage for CA-MRSA in addition to streptococcal species. Failure to recognize resistant organisms or chronic infectious causes can lead to treatment failure and progression of disease.
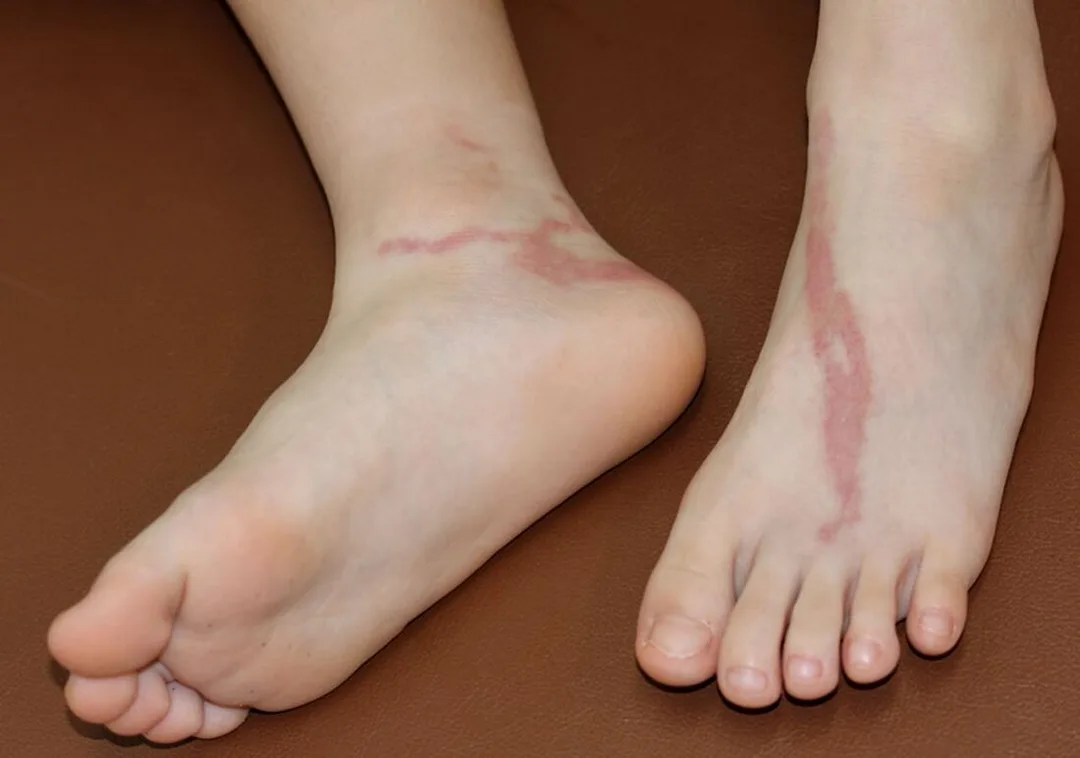
- Published on
Emergency and Acute Medicine – Lymphadenitis
Basics description
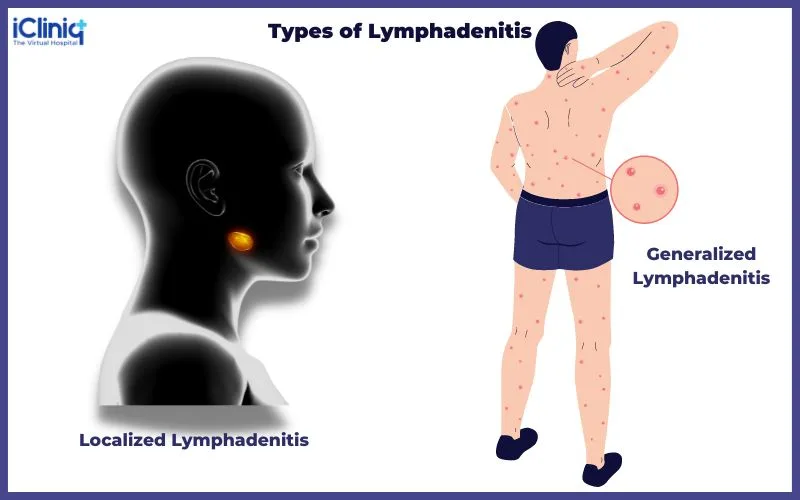
Lymphadenitis refers to inflammation and enlargement of lymph nodes, most commonly as part of a systemic response to infection. Nodes become engorged with lymphocytes and macrophages and may be secondarily involved from infection in a distal extremity, producing painful, tender adenopathy proximally. Acute suppurative lymphadenitis may follow pharyngeal or skin infections and can progress to abscess formation.
Etiology
Lymphadenitis is most frequently caused by bacterial infection. The most common organisms in pyogenic lymphadenitis are Staphylococcus aureus, including community-associated methicillin-resistant S. aureus (CA-MRSA), and group A β-hemolytic Streptococcus. CA-MRSA risk factors include prior MRSA infection, household exposure, military service, incarceration, contact sports, injection drug use, and men who have sex with men. Cervical lymphadenitis usually originates from pharyngeal or periodontal infections and commonly involves streptococci and anaerobes. Axillary lymphadenitis is often caused by group A streptococcus. Nosocomial MRSA should be suspected in patients with recent hospitalization, surgery, dialysis, vascular catheters, recent antibiotic use, or unresponsive infection. In children, acute unilateral cervical suppurative lymphadenitis is most common in those younger than six years and is typically caused by S. aureus, group A streptococcus, or anaerobes.
Diagnosis signs and symptoms
Patients typically present with painful swelling and inflammation of affected lymph nodes, often in association with cellulitis or abscess if the cause is pyogenic. Axillary lymphadenitis may present with fever, axillary pain, and acute lymphedema of the arm or chest and may be associated with ipsilateral pleural effusion. History should include duration of lymphadenopathy, pain, fever, night sweats, weight loss, fatigue, sore throat, cough, occupational and animal exposures, sexual history, drug use, and travel. Physical examination should assess whether lymphadenopathy is localized or generalized, node size, tenderness, overlying skin changes, presence of skin lesions, splenomegaly, and involvement of supraclavicular or scalene nodes, which is always abnormal.
Essential workup
Acute regional lymphadenitis is usually a clinical diagnosis and often part of a broader infectious syndrome such as cellulitis. History and physical examination should focus on identifying an infectious source.
Diagnosis tests and interpretation
Laboratory testing is not always required. A CBC may show leukocytosis with left shift or be normal. Serologic testing for EBV, CMV, HIV, or other pathogens should be guided by clinical suspicion. Ultrasound or CT imaging is indicated when patients fail to improve with therapy or when suppuration is suspected. Percutaneous needle aspiration or surgical drainage should be considered if abscess formation occurs or if there is poor clinical response.
Differential diagnosis
The differential diagnosis includes common infections such as adenovirus, scarlet fever, cat scratch disease, fungal infections, and herpes zoster, as well as unusual infections including sporotrichosis, diphtheria, plague, anthrax, typhoid, rubella, and West Nile virus. Sexually transmitted infections, systemic infections such as HIV, infectious mononucleosis, toxoplasmosis, tuberculosis, hepatitis, and dengue should be considered. Noninfectious causes include drug reactions, malignancy, rheumatologic disorders, and pediatric-specific conditions such as Kawasaki disease and PFAPA syndrome.
Treatment
Initial management includes ensuring airway, breathing, and circulation stability. Treatment is directed at the underlying cause and should account for local resistance patterns, including CA-MRSA prevalence. Outpatient therapy typically lasts 7–10 days and includes limb elevation, moist heat, analgesics, and antibiotics. Abscesses require drainage with culture when possible. Skin-source infections are commonly treated with oral cephalexin plus trimethoprim–sulfamethoxazole or alternatives such as clindamycin or doxycycline. Pharyngeal or periodontal sources are treated with penicillin VK or alternatives such as clindamycin or amoxicillin–clavulanate. Inpatient therapy may require IV penicillin-based regimens with MRSA coverage using vancomycin or clindamycin when indicated.
Disposition and follow-up
Admission is indicated for toxic-appearing patients, those with immunosuppression or significant comorbidities, inability to tolerate oral therapy, or unreliable follow-up. Patients with mild infection who are nontoxic, can take oral antibiotics, and have reliable follow-up within 24–48 hours may be discharged. Failure to resolve promptly with antibiotics should prompt evaluation for malignancy or other serious causes, and lymph node biopsy may be indicated for persistent, large, or supraclavicular nodes.
Pearls and pitfalls
Staphylococcus species are the most common cause of acute regional pyogenic lymphadenitis. Empiric antibiotic therapy should include coverage for CA-MRSA in addition to streptococci, particularly in unresponsive or high-risk infections.
Basics description
Lymphadenitis refers to inflammation and enlargement of lymph nodes, most commonly as part of a systemic response to infection. Nodes become engorged with lymphocytes and macrophages and may be secondarily involved from infection in a distal extremity, producing painful, tender adenopathy proximally. Acute suppurative lymphadenitis may follow pharyngeal or skin infections and can progress to abscess formation.
Etiology
Lymphadenitis is most frequently caused by bacterial infection. The most common organisms in pyogenic lymphadenitis are Staphylococcus aureus, including community-associated methicillin-resistant S. aureus (CA-MRSA), and group A β-hemolytic Streptococcus. CA-MRSA risk factors include prior MRSA infection, household exposure, military service, incarceration, contact sports, injection drug use, and men who have sex with men. Cervical lymphadenitis usually originates from pharyngeal or periodontal infections and commonly involves streptococci and anaerobes. Axillary lymphadenitis is often caused by group A streptococcus. Nosocomial MRSA should be suspected in patients with recent hospitalization, surgery, dialysis, vascular catheters, recent antibiotic use, or unresponsive infection. In children, acute unilateral cervical suppurative lymphadenitis is most common in those younger than six years and is typically caused by S. aureus, group A streptococcus, or anaerobes.
Diagnosis signs and symptoms
Patients typically present with painful swelling and inflammation of affected lymph nodes, often in association with cellulitis or abscess if the cause is pyogenic. Axillary lymphadenitis may present with fever, axillary pain, and acute lymphedema of the arm or chest and may be associated with ipsilateral pleural effusion. History should include duration of lymphadenopathy, pain, fever, night sweats, weight loss, fatigue, sore throat, cough, occupational and animal exposures, sexual history, drug use, and travel. Physical examination should assess whether lymphadenopathy is localized or generalized, node size, tenderness, overlying skin changes, presence of skin lesions, splenomegaly, and involvement of supraclavicular or scalene nodes, which is always abnormal.
Essential workup
Acute regional lymphadenitis is usually a clinical diagnosis and often part of a broader infectious syndrome such as cellulitis. History and physical examination should focus on identifying an infectious source.
Diagnosis tests and interpretation
Laboratory testing is not always required. A CBC may show leukocytosis with left shift or be normal. Serologic testing for EBV, CMV, HIV, or other pathogens should be guided by clinical suspicion. Ultrasound or CT imaging is indicated when patients fail to improve with therapy or when suppuration is suspected. Percutaneous needle aspiration or surgical drainage should be considered if abscess formation occurs or if there is poor clinical response.
Differential diagnosis
The differential diagnosis includes common infections such as adenovirus, scarlet fever, cat scratch disease, fungal infections, and herpes zoster, as well as unusual infections including sporotrichosis, diphtheria, plague, anthrax, typhoid, rubella, and West Nile virus. Sexually transmitted infections, systemic infections such as HIV, infectious mononucleosis, toxoplasmosis, tuberculosis, hepatitis, and dengue should be considered. Noninfectious causes include drug reactions, malignancy, rheumatologic disorders, and pediatric-specific conditions such as Kawasaki disease and PFAPA syndrome.
Treatment
Initial management includes ensuring airway, breathing, and circulation stability. Treatment is directed at the underlying cause and should account for local resistance patterns, including CA-MRSA prevalence. Outpatient therapy typically lasts 7–10 days and includes limb elevation, moist heat, analgesics, and antibiotics. Abscesses require drainage with culture when possible. Skin-source infections are commonly treated with oral cephalexin plus trimethoprim–sulfamethoxazole or alternatives such as clindamycin or doxycycline. Pharyngeal or periodontal sources are treated with penicillin VK or alternatives such as clindamycin or amoxicillin–clavulanate. Inpatient therapy may require IV penicillin-based regimens with MRSA coverage using vancomycin or clindamycin when indicated.
Disposition and follow-up
Admission is indicated for toxic-appearing patients, those with immunosuppression or significant comorbidities, inability to tolerate oral therapy, or unreliable follow-up. Patients with mild infection who are nontoxic, can take oral antibiotics, and have reliable follow-up within 24–48 hours may be discharged. Failure to resolve promptly with antibiotics should prompt evaluation for malignancy or other serious causes, and lymph node biopsy may be indicated for persistent, large, or supraclavicular nodes.
Pearls and pitfalls
Staphylococcus species are the most common cause of acute regional pyogenic lymphadenitis. Empiric antibiotic therapy should include coverage for CA-MRSA in addition to streptococci, particularly in unresponsive or high-risk infections.
- Published on
Emergency and Acute Medicine – Lymphadenitis
Basics description
Lymphadenitis refers to inflammation and enlargement of lymph nodes, most commonly as part of a systemic response to infection. Nodes become engorged with lymphocytes and macrophages and may be secondarily involved from infection in a distal extremity, producing painful, tender adenopathy proximally. Acute suppurative lymphadenitis may follow pharyngeal or skin infections and can progress to abscess formation.
Etiology
Lymphadenitis is most frequently caused by bacterial infection. The most common organisms in pyogenic lymphadenitis are Staphylococcus aureus, including community-associated methicillin-resistant S. aureus (CA-MRSA), and group A β-hemolytic Streptococcus. CA-MRSA risk factors include prior MRSA infection, household exposure, military service, incarceration, contact sports, injection drug use, and men who have sex with men. Cervical lymphadenitis usually originates from pharyngeal or periodontal infections and commonly involves streptococci and anaerobes. Axillary lymphadenitis is often caused by group A streptococcus. Nosocomial MRSA should be suspected in patients with recent hospitalization, surgery, dialysis, vascular catheters, recent antibiotic use, or unresponsive infection. In children, acute unilateral cervical suppurative lymphadenitis is most common in those younger than six years and is typically caused by S. aureus, group A streptococcus, or anaerobes.
Diagnosis signs and symptoms
Patients typically present with painful swelling and inflammation of affected lymph nodes, often in association with cellulitis or abscess if the cause is pyogenic. Axillary lymphadenitis may present with fever, axillary pain, and acute lymphedema of the arm or chest and may be associated with ipsilateral pleural effusion. History should include duration of lymphadenopathy, pain, fever, night sweats, weight loss, fatigue, sore throat, cough, occupational and animal exposures, sexual history, drug use, and travel. Physical examination should assess whether lymphadenopathy is localized or generalized, node size, tenderness, overlying skin changes, presence of skin lesions, splenomegaly, and involvement of supraclavicular or scalene nodes, which is always abnormal.
Essential workup
Acute regional lymphadenitis is usually a clinical diagnosis and often part of a broader infectious syndrome such as cellulitis. History and physical examination should focus on identifying an infectious source.
Diagnosis tests and interpretation
Laboratory testing is not always required. A CBC may show leukocytosis with left shift or be normal. Serologic testing for EBV, CMV, HIV, or other pathogens should be guided by clinical suspicion. Ultrasound or CT imaging is indicated when patients fail to improve with therapy or when suppuration is suspected. Percutaneous needle aspiration or surgical drainage should be considered if abscess formation occurs or if there is poor clinical response.
Differential diagnosis
The differential diagnosis includes common infections such as adenovirus, scarlet fever, cat scratch disease, fungal infections, and herpes zoster, as well as unusual infections including sporotrichosis, diphtheria, plague, anthrax, typhoid, rubella, and West Nile virus. Sexually transmitted infections, systemic infections such as HIV, infectious mononucleosis, toxoplasmosis, tuberculosis, hepatitis, and dengue should be considered. Noninfectious causes include drug reactions, malignancy, rheumatologic disorders, and pediatric-specific conditions such as Kawasaki disease and PFAPA syndrome.
Treatment
Initial management includes ensuring airway, breathing, and circulation stability. Treatment is directed at the underlying cause and should account for local resistance patterns, including CA-MRSA prevalence. Outpatient therapy typically lasts 7–10 days and includes limb elevation, moist heat, analgesics, and antibiotics. Abscesses require drainage with culture when possible. Skin-source infections are commonly treated with oral cephalexin plus trimethoprim–sulfamethoxazole or alternatives such as clindamycin or doxycycline. Pharyngeal or periodontal sources are treated with penicillin VK or alternatives such as clindamycin or amoxicillin–clavulanate. Inpatient therapy may require IV penicillin-based regimens with MRSA coverage using vancomycin or clindamycin when indicated.
Disposition and follow-up
Admission is indicated for toxic-appearing patients, those with immunosuppression or significant comorbidities, inability to tolerate oral therapy, or unreliable follow-up. Patients with mild infection who are nontoxic, can take oral antibiotics, and have reliable follow-up within 24–48 hours may be discharged. Failure to resolve promptly with antibiotics should prompt evaluation for malignancy or other serious causes, and lymph node biopsy may be indicated for persistent, large, or supraclavicular nodes.
Pearls and pitfalls
Staphylococcus species are the most common cause of acute regional pyogenic lymphadenitis. Empiric antibiotic therapy should include coverage for CA-MRSA in addition to streptococci, particularly in unresponsive or high-risk infections.
Basics description
Lymphadenitis refers to inflammation and enlargement of lymph nodes, most commonly as part of a systemic response to infection. Nodes become engorged with lymphocytes and macrophages and may be secondarily involved from infection in a distal extremity, producing painful, tender adenopathy proximally. Acute suppurative lymphadenitis may follow pharyngeal or skin infections and can progress to abscess formation.
Etiology
Lymphadenitis is most frequently caused by bacterial infection. The most common organisms in pyogenic lymphadenitis are Staphylococcus aureus, including community-associated methicillin-resistant S. aureus (CA-MRSA), and group A β-hemolytic Streptococcus. CA-MRSA risk factors include prior MRSA infection, household exposure, military service, incarceration, contact sports, injection drug use, and men who have sex with men. Cervical lymphadenitis usually originates from pharyngeal or periodontal infections and commonly involves streptococci and anaerobes. Axillary lymphadenitis is often caused by group A streptococcus. Nosocomial MRSA should be suspected in patients with recent hospitalization, surgery, dialysis, vascular catheters, recent antibiotic use, or unresponsive infection. In children, acute unilateral cervical suppurative lymphadenitis is most common in those younger than six years and is typically caused by S. aureus, group A streptococcus, or anaerobes.
Diagnosis signs and symptoms
Patients typically present with painful swelling and inflammation of affected lymph nodes, often in association with cellulitis or abscess if the cause is pyogenic. Axillary lymphadenitis may present with fever, axillary pain, and acute lymphedema of the arm or chest and may be associated with ipsilateral pleural effusion. History should include duration of lymphadenopathy, pain, fever, night sweats, weight loss, fatigue, sore throat, cough, occupational and animal exposures, sexual history, drug use, and travel. Physical examination should assess whether lymphadenopathy is localized or generalized, node size, tenderness, overlying skin changes, presence of skin lesions, splenomegaly, and involvement of supraclavicular or scalene nodes, which is always abnormal.
Essential workup
Acute regional lymphadenitis is usually a clinical diagnosis and often part of a broader infectious syndrome such as cellulitis. History and physical examination should focus on identifying an infectious source.
Diagnosis tests and interpretation
Laboratory testing is not always required. A CBC may show leukocytosis with left shift or be normal. Serologic testing for EBV, CMV, HIV, or other pathogens should be guided by clinical suspicion. Ultrasound or CT imaging is indicated when patients fail to improve with therapy or when suppuration is suspected. Percutaneous needle aspiration or surgical drainage should be considered if abscess formation occurs or if there is poor clinical response.
Differential diagnosis
The differential diagnosis includes common infections such as adenovirus, scarlet fever, cat scratch disease, fungal infections, and herpes zoster, as well as unusual infections including sporotrichosis, diphtheria, plague, anthrax, typhoid, rubella, and West Nile virus. Sexually transmitted infections, systemic infections such as HIV, infectious mononucleosis, toxoplasmosis, tuberculosis, hepatitis, and dengue should be considered. Noninfectious causes include drug reactions, malignancy, rheumatologic disorders, and pediatric-specific conditions such as Kawasaki disease and PFAPA syndrome.
Treatment
Initial management includes ensuring airway, breathing, and circulation stability. Treatment is directed at the underlying cause and should account for local resistance patterns, including CA-MRSA prevalence. Outpatient therapy typically lasts 7–10 days and includes limb elevation, moist heat, analgesics, and antibiotics. Abscesses require drainage with culture when possible. Skin-source infections are commonly treated with oral cephalexin plus trimethoprim–sulfamethoxazole or alternatives such as clindamycin or doxycycline. Pharyngeal or periodontal sources are treated with penicillin VK or alternatives such as clindamycin or amoxicillin–clavulanate. Inpatient therapy may require IV penicillin-based regimens with MRSA coverage using vancomycin or clindamycin when indicated.
Disposition and follow-up
Admission is indicated for toxic-appearing patients, those with immunosuppression or significant comorbidities, inability to tolerate oral therapy, or unreliable follow-up. Patients with mild infection who are nontoxic, can take oral antibiotics, and have reliable follow-up within 24–48 hours may be discharged. Failure to resolve promptly with antibiotics should prompt evaluation for malignancy or other serious causes, and lymph node biopsy may be indicated for persistent, large, or supraclavicular nodes.
Pearls and pitfalls
Staphylococcus species are the most common cause of acute regional pyogenic lymphadenitis. Empiric antibiotic therapy should include coverage for CA-MRSA in addition to streptococci, particularly in unresponsive or high-risk infections.
- Published on
KembaraXtra-Medicine – Atrial Septal Defect
An atrial septal defect (ASD) is a congenital or iatrogenic defect in the atrial septum that creates an abnormal communication between the left and right atria, resulting in interatrial shunting of blood. This shunt is typically left to right due to higher left atrial pressures, leading to increased pulmonary blood flow and right-sided volume overload. ASDs are distinct from a patent foramen ovale (PFO), which represents postnatal failure of closure of a normal fetal structure rather than a true defect in septal formation. PFOs occur in approximately 20% to 25% of the adult population, whereas ASDs represent abnormal septal development established in utero.
ASDs are classified based on their anatomic location. Ostium primum defects occur in the inferior portion of the atrial septum due to failure of fusion of the septum primum with the endocardial cushions and are often associated with a cleft anterior mitral leaflet. Ostium secundum defects are the most common type and result from incomplete fusion of the septum primum and septum secundum, usually near the fossa ovalis. Sinus venosus defects occur near the junction of the right atrium with the superior or inferior vena cava and are frequently associated with anomalous pulmonary venous return. Coronary sinus septal defects, also known as unroofed coronary sinus, involve abnormal communication between the coronary sinus and the left atrium and are often associated with a persistent left superior vena cava. Iatrogenic ASDs may occur following procedures requiring transseptal puncture, such as electrophysiologic or structural heart interventions.
ASDs are among the most common acyanotic congenital heart diseases, occurring in approximately 0.1% of live births and accounting for 30% to 40% of clinically significant intracardiac shunts in adults. Ostium secundum defects account for about 75% of cases, followed by ostium primum defects (15%–20%), sinus venosus defects (5%–10%), and coronary sinus defects (<1%). secundum asds are more common in females, whereas gender distribution is similar other types. may occur isolation or association with genetic syndromes such as down syndrome, holt-oram noonan and digeorge well congenital cardiac abnormalities.< />pan>
Most ASDs are small and asymptomatic during infancy and childhood, but many patients develop symptoms later in life regardless of defect size. Exertional dyspnea and fatigue are the most common presenting symptoms. Small ASDs are often detected incidentally due to a cardiac murmur on routine examination. Large defects in infancy may lead to heart failure, recurrent respiratory infections, or failure to thrive. In adults, complications include atrial arrhythmias such as atrial fibrillation or flutter, paradoxical embolism causing ischemic stroke, and rarely platypnea-orthodeoxia syndrome. Physical examination findings may include right ventricular lift, wide fixed splitting of the second heart sound, systolic ejection murmurs due to increased pulmonary flow, and diastolic tricuspid flow murmurs in the presence of large shunts.
Diagnosis is based on clinical evaluation and imaging. Electrocardiography may show right axis deviation, right bundle branch block, atrial enlargement, or atrial arrhythmias depending on the ASD type. Chest radiography often demonstrates cardiomegaly, right-sided chamber enlargement, and increased pulmonary vascular markings. Transthoracic echocardiography is the primary diagnostic tool and is highly sensitive for secundum and primum defects, particularly when subcostal views and color Doppler imaging are used. Transesophageal echocardiography provides near-complete sensitivity and specificity and is essential when device closure is being considered, allowing accurate assessment of defect size and surrounding rims. Cardiac catheterization is used to quantify shunt magnitude (Qp:Qs), assess pulmonary vascular resistance, and evaluate pulmonary hypertension. Cardiac MRI and CT are valuable when echocardiography is inconclusive and for detailed assessment of right ventricular size, function, and anomalous pulmonary venous connections.
Management depends on patient age, symptoms, shunt magnitude, right-sided chamber enlargement, and pulmonary vascular status. In adults, ASD closure is recommended when there is significant left-to-right shunting (Qp:Qs >1.5:1), right atrial or ventricular enlargement, symptoms, or pulmonary hypertension that is not advanced or irreversible. Closure is also reasonable for paradoxical embolism or documented platypnea-orthodeoxia. Closure is contraindicated in patients with severe pulmonary hypertension, high pulmonary vascular resistance, or net right-to-left shunting. In children, closure is generally recommended before age 10 when significant shunting or right-sided dilation is present.
Percutaneous device closure is the preferred approach for suitable secundum ASDs and has a success rate exceeding 95% in appropriately selected patients. Surgical closure is indicated for primum, sinus venosus, and coronary sinus defects, or when anatomy is unsuitable for device closure. Iatrogenic ASDs often close spontaneously but may require intervention if associated with significant shunting or complications. Post-closure follow-up includes antiplatelet therapy for several months, serial echocardiography, and monitoring for arrhythmias or device-related complications.
Untreated large ASDs are associated with increased mortality and long-term complications, including right heart failure, pulmonary arterial hypertension, Eisenmenger syndrome, atrial arrhythmias, and embolic events. Patients with small shunts generally have a normal life expectancy but require periodic monitoring. ASD repair improves symptoms, exercise capacity, quality of life, and long-term survival, particularly when performed earlier in life. Long-term follow-up is recommended after repair, especially in adults, to monitor for arrhythmias, pulmonary hypertension, ventricular dysfunction, and residual shunts.
An atrial septal defect (ASD) is a congenital or iatrogenic defect in the atrial septum that creates an abnormal communication between the left and right atria, resulting in interatrial shunting of blood. This shunt is typically left to right due to higher left atrial pressures, leading to increased pulmonary blood flow and right-sided volume overload. ASDs are distinct from a patent foramen ovale (PFO), which represents postnatal failure of closure of a normal fetal structure rather than a true defect in septal formation. PFOs occur in approximately 20% to 25% of the adult population, whereas ASDs represent abnormal septal development established in utero.
ASDs are classified based on their anatomic location. Ostium primum defects occur in the inferior portion of the atrial septum due to failure of fusion of the septum primum with the endocardial cushions and are often associated with a cleft anterior mitral leaflet. Ostium secundum defects are the most common type and result from incomplete fusion of the septum primum and septum secundum, usually near the fossa ovalis. Sinus venosus defects occur near the junction of the right atrium with the superior or inferior vena cava and are frequently associated with anomalous pulmonary venous return. Coronary sinus septal defects, also known as unroofed coronary sinus, involve abnormal communication between the coronary sinus and the left atrium and are often associated with a persistent left superior vena cava. Iatrogenic ASDs may occur following procedures requiring transseptal puncture, such as electrophysiologic or structural heart interventions.
ASDs are among the most common acyanotic congenital heart diseases, occurring in approximately 0.1% of live births and accounting for 30% to 40% of clinically significant intracardiac shunts in adults. Ostium secundum defects account for about 75% of cases, followed by ostium primum defects (15%–20%), sinus venosus defects (5%–10%), and coronary sinus defects (<1%). secundum asds are more common in females, whereas gender distribution is similar other types. may occur isolation or association with genetic syndromes such as down syndrome, holt-oram noonan and digeorge well congenital cardiac abnormalities.< />pan>
Most ASDs are small and asymptomatic during infancy and childhood, but many patients develop symptoms later in life regardless of defect size. Exertional dyspnea and fatigue are the most common presenting symptoms. Small ASDs are often detected incidentally due to a cardiac murmur on routine examination. Large defects in infancy may lead to heart failure, recurrent respiratory infections, or failure to thrive. In adults, complications include atrial arrhythmias such as atrial fibrillation or flutter, paradoxical embolism causing ischemic stroke, and rarely platypnea-orthodeoxia syndrome. Physical examination findings may include right ventricular lift, wide fixed splitting of the second heart sound, systolic ejection murmurs due to increased pulmonary flow, and diastolic tricuspid flow murmurs in the presence of large shunts.
Diagnosis is based on clinical evaluation and imaging. Electrocardiography may show right axis deviation, right bundle branch block, atrial enlargement, or atrial arrhythmias depending on the ASD type. Chest radiography often demonstrates cardiomegaly, right-sided chamber enlargement, and increased pulmonary vascular markings. Transthoracic echocardiography is the primary diagnostic tool and is highly sensitive for secundum and primum defects, particularly when subcostal views and color Doppler imaging are used. Transesophageal echocardiography provides near-complete sensitivity and specificity and is essential when device closure is being considered, allowing accurate assessment of defect size and surrounding rims. Cardiac catheterization is used to quantify shunt magnitude (Qp:Qs), assess pulmonary vascular resistance, and evaluate pulmonary hypertension. Cardiac MRI and CT are valuable when echocardiography is inconclusive and for detailed assessment of right ventricular size, function, and anomalous pulmonary venous connections.
Management depends on patient age, symptoms, shunt magnitude, right-sided chamber enlargement, and pulmonary vascular status. In adults, ASD closure is recommended when there is significant left-to-right shunting (Qp:Qs >1.5:1), right atrial or ventricular enlargement, symptoms, or pulmonary hypertension that is not advanced or irreversible. Closure is also reasonable for paradoxical embolism or documented platypnea-orthodeoxia. Closure is contraindicated in patients with severe pulmonary hypertension, high pulmonary vascular resistance, or net right-to-left shunting. In children, closure is generally recommended before age 10 when significant shunting or right-sided dilation is present.
Percutaneous device closure is the preferred approach for suitable secundum ASDs and has a success rate exceeding 95% in appropriately selected patients. Surgical closure is indicated for primum, sinus venosus, and coronary sinus defects, or when anatomy is unsuitable for device closure. Iatrogenic ASDs often close spontaneously but may require intervention if associated with significant shunting or complications. Post-closure follow-up includes antiplatelet therapy for several months, serial echocardiography, and monitoring for arrhythmias or device-related complications.
Untreated large ASDs are associated with increased mortality and long-term complications, including right heart failure, pulmonary arterial hypertension, Eisenmenger syndrome, atrial arrhythmias, and embolic events. Patients with small shunts generally have a normal life expectancy but require periodic monitoring. ASD repair improves symptoms, exercise capacity, quality of life, and long-term survival, particularly when performed earlier in life. Long-term follow-up is recommended after repair, especially in adults, to monitor for arrhythmias, pulmonary hypertension, ventricular dysfunction, and residual shunts.
- Published on
KembaraXtra-Medicine – Atrioventricular Dissociation
Atrioventricular (AV) dissociation means there is no consistent relationship between atrial activity and ventricular activity—the atria and ventricles are functioning independently. This is an umbrella concept rather than a single diagnosis, because AV dissociation can appear in several different rhythm problems, including slow rhythms (bradycardias), complete heart block, and fast rhythms (tachycardias) such as ventricular tachycardia or situations where an atrial rhythm coexists with an accelerated junctional rhythm or AV nodal reentrant tachycardia.
AV dissociation is sometimes referred to as complete AV block or third-degree AV block, and it is associated with the ICD-10CM code I44.2 (Atrioventricular block, complete). Its overall “prevalence” depends on how common the underlying conditions are that produce AV dissociation, rather than AV dissociation being counted as one separate disease on its own.
Clinical findings can be normal if the rhythm is not causing problems with blood flow. If the right atrium contracts against a closed tricuspid valve during ventricular systole, cannon A waves may be visible in the jugular venous pulse. Symptoms vary depending on the rhythm and the patient’s stability and may include dizziness, palpitations, syncope or presyncope (from reduced cardiac output), fatigue and reduced exercise tolerance, mental status changes, congestive heart failure symptoms, or angina. Some patients may have no symptoms at all.
Causes of AV dissociation include a sinus node that fires too slowly, or a ventricular/junctional pacemaker that is firing inappropriately fast relative to the atria. It can also be iatrogenic, such as from anesthesia, inotrope infusions, ventricular pacing, radiofrequency ablation (for example, slow pathway ablation), or digoxin toxicity. Other contributors include sinus node disease, ischemia, hyperkalemia, and high vagal tone. When AV dissociation occurs due to complete heart block, causes include progressive fibrosis of the His–Purkinje system, medications, and infections such as Lyme disease.
Diagnosis is based on ECG evidence of atrial and ventricular activity that are not linked in a consistent pattern. The differential diagnosis should focus on rhythm disorders that can produce AV dissociation. Importantly, the atrial rate does not have to be faster than the ventricular rate for AV dissociation (that “atrial faster than ventricular” idea fits more specifically with the classic definition of complete heart block). Two related patterns include isorhythmic AV dissociation, where atrial and ventricular rates are similar but dissociated, and interference dissociation, where atrial and ventricular rates are close and occasional conduction may occur.
Workup should be guided by the clinical situation. Routine labs, cardiac biomarkers, and imaging may be needed depending on symptoms and suspected cause, with special attention to electrolytes (especially potassium) and a digoxin level when relevant. If complete heart block is suspected and exposure risk is plausible, Lyme antibody testing should be considered.
Treatment depends first on whether the patient is stable and on whether the rhythm is slow or fast. In bradycardic AV dissociation with symptoms or hemodynamic compromise, a temporary pacemaker is the most reliable immediate therapy. AV nodal blocking agents should be held, and chronotropic medications such as atropine, dopamine, dobutamine, or isoproterenol can be used as temporary measures while preparing for pacing when appropriate. In tachycardic causes such as ventricular tachycardia, unstable patients should receive cardioversion first. Intravenous antiarrhythmics such as amiodarone or lidocaine may be used to suppress the arrhythmia, and definitive management focuses on the underlying cause, such as evaluating ischemia (including coronary angiography if indicated) or electrophysiology study with possible ablation.
All patients with AV dissociation should be referred to a cardiologist for evaluation of the rhythm and its cause. The key reminder is that AV dissociation itself is a sign/pattern—management, prognosis, and disposition are determined by the specific arrhythmia and clinical context producing it.
Atrioventricular (AV) dissociation means there is no consistent relationship between atrial activity and ventricular activity—the atria and ventricles are functioning independently. This is an umbrella concept rather than a single diagnosis, because AV dissociation can appear in several different rhythm problems, including slow rhythms (bradycardias), complete heart block, and fast rhythms (tachycardias) such as ventricular tachycardia or situations where an atrial rhythm coexists with an accelerated junctional rhythm or AV nodal reentrant tachycardia.
AV dissociation is sometimes referred to as complete AV block or third-degree AV block, and it is associated with the ICD-10CM code I44.2 (Atrioventricular block, complete). Its overall “prevalence” depends on how common the underlying conditions are that produce AV dissociation, rather than AV dissociation being counted as one separate disease on its own.
Clinical findings can be normal if the rhythm is not causing problems with blood flow. If the right atrium contracts against a closed tricuspid valve during ventricular systole, cannon A waves may be visible in the jugular venous pulse. Symptoms vary depending on the rhythm and the patient’s stability and may include dizziness, palpitations, syncope or presyncope (from reduced cardiac output), fatigue and reduced exercise tolerance, mental status changes, congestive heart failure symptoms, or angina. Some patients may have no symptoms at all.
Causes of AV dissociation include a sinus node that fires too slowly, or a ventricular/junctional pacemaker that is firing inappropriately fast relative to the atria. It can also be iatrogenic, such as from anesthesia, inotrope infusions, ventricular pacing, radiofrequency ablation (for example, slow pathway ablation), or digoxin toxicity. Other contributors include sinus node disease, ischemia, hyperkalemia, and high vagal tone. When AV dissociation occurs due to complete heart block, causes include progressive fibrosis of the His–Purkinje system, medications, and infections such as Lyme disease.
Diagnosis is based on ECG evidence of atrial and ventricular activity that are not linked in a consistent pattern. The differential diagnosis should focus on rhythm disorders that can produce AV dissociation. Importantly, the atrial rate does not have to be faster than the ventricular rate for AV dissociation (that “atrial faster than ventricular” idea fits more specifically with the classic definition of complete heart block). Two related patterns include isorhythmic AV dissociation, where atrial and ventricular rates are similar but dissociated, and interference dissociation, where atrial and ventricular rates are close and occasional conduction may occur.
Workup should be guided by the clinical situation. Routine labs, cardiac biomarkers, and imaging may be needed depending on symptoms and suspected cause, with special attention to electrolytes (especially potassium) and a digoxin level when relevant. If complete heart block is suspected and exposure risk is plausible, Lyme antibody testing should be considered.
Treatment depends first on whether the patient is stable and on whether the rhythm is slow or fast. In bradycardic AV dissociation with symptoms or hemodynamic compromise, a temporary pacemaker is the most reliable immediate therapy. AV nodal blocking agents should be held, and chronotropic medications such as atropine, dopamine, dobutamine, or isoproterenol can be used as temporary measures while preparing for pacing when appropriate. In tachycardic causes such as ventricular tachycardia, unstable patients should receive cardioversion first. Intravenous antiarrhythmics such as amiodarone or lidocaine may be used to suppress the arrhythmia, and definitive management focuses on the underlying cause, such as evaluating ischemia (including coronary angiography if indicated) or electrophysiology study with possible ablation.
All patients with AV dissociation should be referred to a cardiologist for evaluation of the rhythm and its cause. The key reminder is that AV dissociation itself is a sign/pattern—management, prognosis, and disposition are determined by the specific arrhythmia and clinical context producing it.

- Published on
KembaraXtra-Medicine – Attention-Deficit/Hyperactivity Disorder
Attention-deficit/hyperactivity disorder (ADHD) is a chronic neurodevelopmental condition characterized by persistent patterns of inattention and/or hyperactivity–impulsivity. Symptoms begin in childhood, persist for at least six months, and cause functional impairment across multiple settings such as home, school, or social environments. ADHD is also referred to as attention deficit disorder (ADD), hyperactivity, or attention deficit syndrome with hyperactivity. Diagnostic classifications include ICD-11 codes (6A05 series) and DSM-5 presentations that are predominantly inattentive, predominantly hyperactive-impulsive, or combined.
ADHD is the most common neurodevelopmental disorder in children, affecting approximately 9%–15% of school-age children, up to 14% of adolescents, and 3%–5% of adults. It is diagnosed more often in males during childhood, particularly in the hyperactive type, while adult prevalence is closer to equal between sexes. Symptoms must be present before age 12, commonly leading to diagnosis between ages 6 and 9. Although hyperactivity may decrease with age, many individuals continue to experience symptoms into adolescence and adulthood.
Risk factors include genetic predisposition, prenatal exposure to tobacco, alcohol, or illicit substances, prematurity, low birth weight, lead exposure, head trauma in early childhood, and psychosocial stressors such as family dysfunction or low socioeconomic status. Genetics play a major role, with first-degree relatives having a two- to sixfold increased risk. Neuroimaging studies show structural and functional brain differences, particularly in frontal and prefrontal regions, although these findings are not diagnostic.
Clinically, ADHD presents in three patterns. The predominantly inattentive type involves difficulty organizing, sustaining attention, remembering tasks, and completing work. The predominantly hyperactive-impulsive type features restlessness, excessive talking, impulsivity, and difficulty waiting or inhibiting responses. The combined type includes features of both. ADHD commonly coexists with other conditions such as learning disabilities, oppositional defiant disorder, autism spectrum disorder, anxiety, depression, and substance use disorders. In adults, overt hyperactivity is less common, while restlessness, disorganization, and difficulty completing tasks predominate.
The etiology of ADHD is multifactorial, with strong genetic influences and contributions from altered catecholamine metabolism, delayed cortical maturation, disrupted reward processing, and environmental factors. Dopamine and norepinephrine pathways in the prefrontal cortex are central to attention, executive function, and impulse control and are key to ADHD pathophysiology.
Diagnosis is clinical and based on a thorough history and assessment of symptoms across settings. Differential diagnoses include medical conditions (e.g., hearing or vision impairment, thyroid disease, seizures), psychiatric disorders (e.g., mood disorders, anxiety, autism spectrum disorder), and psychosocial factors. Evaluation should include collateral information from parents, teachers, or partners, as well as the use of ADHD-specific rating scales. Laboratory tests and imaging are not routinely required unless indicated by history or examination.
Treatment is individualized and often multimodal. Pharmacologic therapy, particularly stimulant medications, is the mainstay and has the strongest evidence for symptom control. Stimulants act by increasing dopamine and norepinephrine availability and are available in multiple short- and long-acting formulations. Atomoxetine is a nonstimulant alternative, typically used when stimulants are ineffective or contraindicated. Other options include antidepressants and alpha-2 adrenergic agonists such as clonidine and guanfacine. Medications require regular monitoring for side effects, including cardiovascular effects, appetite suppression, sleep disturbance, and mood changes.
Nonpharmacologic interventions include behavioral therapy, parent training, educational accommodations, organizational skills training, and social skills interventions. These approaches are particularly important for younger children, mild cases, or when comorbid conditions are present. Educational support through individualized plans or 504 accommodations is recommended. Digital therapeutics and structured behavioral strategies may provide additional benefit, while elimination diets and psychotherapy alone are generally not effective for core ADHD symptoms.
ADHD is often a lifelong condition with symptoms persisting into adulthood in a substantial proportion of patients. Long-term risks include academic and occupational difficulties, psychiatric comorbidities, substance use disorders, and impaired social functioning. Referral to a specialist is recommended when diagnosis is uncertain, comorbidities are complex, or response to first-line treatments is inadequate. Early recognition and appropriate treatment improve functional outcomes and quality of life.
Attention-deficit/hyperactivity disorder (ADHD) is a chronic neurodevelopmental condition characterized by persistent patterns of inattention and/or hyperactivity–impulsivity. Symptoms begin in childhood, persist for at least six months, and cause functional impairment across multiple settings such as home, school, or social environments. ADHD is also referred to as attention deficit disorder (ADD), hyperactivity, or attention deficit syndrome with hyperactivity. Diagnostic classifications include ICD-11 codes (6A05 series) and DSM-5 presentations that are predominantly inattentive, predominantly hyperactive-impulsive, or combined.
ADHD is the most common neurodevelopmental disorder in children, affecting approximately 9%–15% of school-age children, up to 14% of adolescents, and 3%–5% of adults. It is diagnosed more often in males during childhood, particularly in the hyperactive type, while adult prevalence is closer to equal between sexes. Symptoms must be present before age 12, commonly leading to diagnosis between ages 6 and 9. Although hyperactivity may decrease with age, many individuals continue to experience symptoms into adolescence and adulthood.
Risk factors include genetic predisposition, prenatal exposure to tobacco, alcohol, or illicit substances, prematurity, low birth weight, lead exposure, head trauma in early childhood, and psychosocial stressors such as family dysfunction or low socioeconomic status. Genetics play a major role, with first-degree relatives having a two- to sixfold increased risk. Neuroimaging studies show structural and functional brain differences, particularly in frontal and prefrontal regions, although these findings are not diagnostic.
Clinically, ADHD presents in three patterns. The predominantly inattentive type involves difficulty organizing, sustaining attention, remembering tasks, and completing work. The predominantly hyperactive-impulsive type features restlessness, excessive talking, impulsivity, and difficulty waiting or inhibiting responses. The combined type includes features of both. ADHD commonly coexists with other conditions such as learning disabilities, oppositional defiant disorder, autism spectrum disorder, anxiety, depression, and substance use disorders. In adults, overt hyperactivity is less common, while restlessness, disorganization, and difficulty completing tasks predominate.
The etiology of ADHD is multifactorial, with strong genetic influences and contributions from altered catecholamine metabolism, delayed cortical maturation, disrupted reward processing, and environmental factors. Dopamine and norepinephrine pathways in the prefrontal cortex are central to attention, executive function, and impulse control and are key to ADHD pathophysiology.
Diagnosis is clinical and based on a thorough history and assessment of symptoms across settings. Differential diagnoses include medical conditions (e.g., hearing or vision impairment, thyroid disease, seizures), psychiatric disorders (e.g., mood disorders, anxiety, autism spectrum disorder), and psychosocial factors. Evaluation should include collateral information from parents, teachers, or partners, as well as the use of ADHD-specific rating scales. Laboratory tests and imaging are not routinely required unless indicated by history or examination.
Treatment is individualized and often multimodal. Pharmacologic therapy, particularly stimulant medications, is the mainstay and has the strongest evidence for symptom control. Stimulants act by increasing dopamine and norepinephrine availability and are available in multiple short- and long-acting formulations. Atomoxetine is a nonstimulant alternative, typically used when stimulants are ineffective or contraindicated. Other options include antidepressants and alpha-2 adrenergic agonists such as clonidine and guanfacine. Medications require regular monitoring for side effects, including cardiovascular effects, appetite suppression, sleep disturbance, and mood changes.
Nonpharmacologic interventions include behavioral therapy, parent training, educational accommodations, organizational skills training, and social skills interventions. These approaches are particularly important for younger children, mild cases, or when comorbid conditions are present. Educational support through individualized plans or 504 accommodations is recommended. Digital therapeutics and structured behavioral strategies may provide additional benefit, while elimination diets and psychotherapy alone are generally not effective for core ADHD symptoms.
ADHD is often a lifelong condition with symptoms persisting into adulthood in a substantial proportion of patients. Long-term risks include academic and occupational difficulties, psychiatric comorbidities, substance use disorders, and impaired social functioning. Referral to a specialist is recommended when diagnosis is uncertain, comorbidities are complex, or response to first-line treatments is inadequate. Early recognition and appropriate treatment improve functional outcomes and quality of life.
- Published on
KembaraXtra-Medicine – Autism Spectrum Disorder
Autism spectrum disorder (ASD) is a biologically based neurodevelopmental disorder that includes a range of developmental disabilities. It is defined by early-appearing social-communication difficulties and restricted, repetitive patterns of behavior, interests, or activities. ASD describes a constellation of social communication deficits and repetitive sensorimotor behaviors, often with a strong genetic component and sometimes other identifiable causes. Outcomes today are generally better than decades ago, with more individuals able to communicate, learn, and live in the community, though many still require ongoing support into adulthood. Clinicians play an important role by helping families access evaluations, referrals, and community resources, and by anticipating major transitions such as starting school and moving into adult services.
Diagnosis is based on DSM-5-TR or ICD-11 criteria. DSM-5-TR requires persistent deficits in social communication and social interaction across multiple contexts (including social-emotional reciprocity, nonverbal communication, and relationships) plus restricted/repetitive patterns of behavior (such as stereotyped movements or speech, insistence on sameness, restricted interests, and sensory hyper- or hyporeactivity). ASD can be further described by whether intellectual disability, language impairment, medical/genetic conditions, or catatonia are present. Comorbid intellectual disability, neurologic/medical problems, and psychiatric disorders are common, and symptom severity varies widely between individuals.
ASD affects an estimated 1% to 3% of children in the United States, with prevalence often described as approximately 1 in 40 children. Rates have increased over recent decades, but it is unclear whether this reflects broader criteria, increased awareness and diagnostic accuracy, or a true rise in frequency. ASD is more common in males, with an estimated male-to-female ratio of about 3:1. Most children are identified by age 4, often because of delayed communication milestones, though presentation and timing of diagnosis can vary depending on language, cognition, and adaptive functioning.
Risk factors include several prenatal and perinatal factors such as hypoxia-related obstetric complications, prenatal infections, maternal use of certain medications (notably valproic acid), maternal health conditions (diabetes, hypertension, obesity, preeclampsia), advanced parental age, multiple gestation, prematurity, and congenital sensory deficits. Environmental exposures have also been associated in some studies, including significant air pollution exposure during pregnancy and early life and heavy maternal smoking. ASD is highly heritable, with an estimated heritability around 80%. Concordance rates rise with genetic relatedness, and research has identified many genetic contributors including polygenic risk and rare variants such as copy number variants. Neurobiologic studies show brain differences in some individuals with ASD, including atypical connectivity and cortical structural differences, but these findings are not diagnostic.
Clinically, ASD is often described by a triad: impairment in social interaction, atypical verbal and nonverbal communication, and repetitive or unusual behaviors. Social difficulties can include poor social-emotional reciprocity and reduced shared attention. Communication may be affected through limited gestures, reduced facial expression, or difficulty using and interpreting nonverbal signals. Repetitive features may include stereotyped movements (such as hand flapping or rocking), repetitive speech (including echolalia), and strong preferences for sameness or routine. Sensory differences are common, including hypersensitivity to sound, touch, or smells, or hyposensitivity such as unusually high pain tolerance. Catatonia can appear in up to 20% of adolescents and adults with ASD.
Many conditions and syndromes are associated with ASD. Approximately 20% to 50% of individuals have intellectual disability, about 50% have ADHD, around 25% have an associated genetic syndrome, and roughly 12% have epilepsy. Examples of associated syndromes include fragile X syndrome, tuberous sclerosis, Angelman syndrome, Rett syndrome, Down syndrome, and DiGeorge syndrome, among others.
The differential diagnosis includes other psychiatric and neurodevelopmental disorders that may share overlapping features, such as ADHD, Tourette syndrome, selective mutism, catatonia, social anxiety, obsessive-compulsive disorder, language disorders, stereotypic movement disorder, and intellectual disability. Social (pragmatic) communication disorder is an important distinction because it involves social communication deficits without the restricted/repetitive behaviors required for ASD. Attachment disorders can also mimic aspects of ASD, particularly in children with histories of early neglect.
Workup focuses on confirming diagnosis and identifying contributing or associated medical conditions, including genetic syndromes. “Red flags” in early social communication should prompt evaluation, such as no vocalizations by 6 months, no consonant babbling by 12 months, no gestures by 12 months, no spontaneous single words by 16 months, no spontaneous phrases by 24 months, or any loss of previously acquired social-communication skills. The American Academy of Pediatrics and the CDC recommend formal ASD screening at 18 and 24 months. Gold-standard assessment includes detailed developmental and family history, evaluation of intellectual/developmental functioning, direct assessment of ASD symptoms, and measurement of adaptive functioning, with additional language, neuropsychologic, motor, or psychiatric assessments as needed.
Laboratory testing may include newborn screening review (such as PKU), lead screening, hearing assessment, and genetic testing (karyotype, chromosome microarray, and targeted DNA testing when indicated). Creatine kinase and TSH may be considered when motor concerns are present. An EEG is recommended when seizures are suspected or when there is language regression. Brain imaging is recommended if macrocephaly, microcephaly, or abnormal tone/motor findings are present.
Treatment emphasizes early and structured intervention. Nonpharmacologic therapy includes behavioral programs at home and school, applied behavioral analysis (ABA) approaches (such as Discrete Trial Training), and development-focused ABA models (such as Pivotal Response Training, Floortime, and the Early Start Denver Model). Specialized educational approaches focusing on communication and life skills (such as TEACCH) are often helpful. Cognitive-behavioral therapy can reduce anxiety in higher-functioning individuals. Family and teacher education and highly structured environments support skill-building and daily functioning.
Medication does not “cure” ASD, but can reduce specific target symptoms. Risperidone and aripiprazole are FDA-approved for managing irritability associated with ASD. Other medications are used off-label depending on symptoms, including SSRIs or atypical antipsychotics for obsessive or ritualistic behaviors, atypical antipsychotics and other agents for aggression or self-injury, stimulants or alpha-2 agonists for hyperactivity/inattention, and SSRIs or other agents for anxiety or depression. Catatonia is treated with lorazepam and sometimes electroconvulsive therapy, and antipsychotics should be avoided in catatonia because they can worsen it. Complementary approaches have limited evidence overall, though melatonin has preliminary evidence for sleep difficulties.
Most individuals with ASD require some level of support as adults. DSM-5-TR describes severity levels based on support needs, from Level 1 (requiring support) to Level 3 (requiring very substantial support). Better outcomes are linked to early identification and intervention, development of functional spoken language, and capacity for inclusion with typical peers. Poorer outcomes are associated with lack of joint attention by age 4, absence of functional speech by age 5, intellectual disability, seizures, significant comorbid medical or psychiatric conditions, and pervasive social disengagement. Referrals may involve specialists for diagnosis (developmental pediatrics, child psychiatry, psychology, neurology, genetics) and therapy services (speech-language therapy, occupational therapy, behavioral therapy), as well as support for caregivers and school planning.
There is no scientific evidence linking childhood vaccination to the development of ASD. Many individuals with ASD have increased rates of medical issues such as sleep problems, gastrointestinal concerns, and oral health problems, and psychiatric comorbidities are common across the lifespan.
Autism spectrum disorder (ASD) is a biologically based neurodevelopmental disorder that includes a range of developmental disabilities. It is defined by early-appearing social-communication difficulties and restricted, repetitive patterns of behavior, interests, or activities. ASD describes a constellation of social communication deficits and repetitive sensorimotor behaviors, often with a strong genetic component and sometimes other identifiable causes. Outcomes today are generally better than decades ago, with more individuals able to communicate, learn, and live in the community, though many still require ongoing support into adulthood. Clinicians play an important role by helping families access evaluations, referrals, and community resources, and by anticipating major transitions such as starting school and moving into adult services.
Diagnosis is based on DSM-5-TR or ICD-11 criteria. DSM-5-TR requires persistent deficits in social communication and social interaction across multiple contexts (including social-emotional reciprocity, nonverbal communication, and relationships) plus restricted/repetitive patterns of behavior (such as stereotyped movements or speech, insistence on sameness, restricted interests, and sensory hyper- or hyporeactivity). ASD can be further described by whether intellectual disability, language impairment, medical/genetic conditions, or catatonia are present. Comorbid intellectual disability, neurologic/medical problems, and psychiatric disorders are common, and symptom severity varies widely between individuals.
ASD affects an estimated 1% to 3% of children in the United States, with prevalence often described as approximately 1 in 40 children. Rates have increased over recent decades, but it is unclear whether this reflects broader criteria, increased awareness and diagnostic accuracy, or a true rise in frequency. ASD is more common in males, with an estimated male-to-female ratio of about 3:1. Most children are identified by age 4, often because of delayed communication milestones, though presentation and timing of diagnosis can vary depending on language, cognition, and adaptive functioning.
Risk factors include several prenatal and perinatal factors such as hypoxia-related obstetric complications, prenatal infections, maternal use of certain medications (notably valproic acid), maternal health conditions (diabetes, hypertension, obesity, preeclampsia), advanced parental age, multiple gestation, prematurity, and congenital sensory deficits. Environmental exposures have also been associated in some studies, including significant air pollution exposure during pregnancy and early life and heavy maternal smoking. ASD is highly heritable, with an estimated heritability around 80%. Concordance rates rise with genetic relatedness, and research has identified many genetic contributors including polygenic risk and rare variants such as copy number variants. Neurobiologic studies show brain differences in some individuals with ASD, including atypical connectivity and cortical structural differences, but these findings are not diagnostic.
Clinically, ASD is often described by a triad: impairment in social interaction, atypical verbal and nonverbal communication, and repetitive or unusual behaviors. Social difficulties can include poor social-emotional reciprocity and reduced shared attention. Communication may be affected through limited gestures, reduced facial expression, or difficulty using and interpreting nonverbal signals. Repetitive features may include stereotyped movements (such as hand flapping or rocking), repetitive speech (including echolalia), and strong preferences for sameness or routine. Sensory differences are common, including hypersensitivity to sound, touch, or smells, or hyposensitivity such as unusually high pain tolerance. Catatonia can appear in up to 20% of adolescents and adults with ASD.
Many conditions and syndromes are associated with ASD. Approximately 20% to 50% of individuals have intellectual disability, about 50% have ADHD, around 25% have an associated genetic syndrome, and roughly 12% have epilepsy. Examples of associated syndromes include fragile X syndrome, tuberous sclerosis, Angelman syndrome, Rett syndrome, Down syndrome, and DiGeorge syndrome, among others.
The differential diagnosis includes other psychiatric and neurodevelopmental disorders that may share overlapping features, such as ADHD, Tourette syndrome, selective mutism, catatonia, social anxiety, obsessive-compulsive disorder, language disorders, stereotypic movement disorder, and intellectual disability. Social (pragmatic) communication disorder is an important distinction because it involves social communication deficits without the restricted/repetitive behaviors required for ASD. Attachment disorders can also mimic aspects of ASD, particularly in children with histories of early neglect.
Workup focuses on confirming diagnosis and identifying contributing or associated medical conditions, including genetic syndromes. “Red flags” in early social communication should prompt evaluation, such as no vocalizations by 6 months, no consonant babbling by 12 months, no gestures by 12 months, no spontaneous single words by 16 months, no spontaneous phrases by 24 months, or any loss of previously acquired social-communication skills. The American Academy of Pediatrics and the CDC recommend formal ASD screening at 18 and 24 months. Gold-standard assessment includes detailed developmental and family history, evaluation of intellectual/developmental functioning, direct assessment of ASD symptoms, and measurement of adaptive functioning, with additional language, neuropsychologic, motor, or psychiatric assessments as needed.
Laboratory testing may include newborn screening review (such as PKU), lead screening, hearing assessment, and genetic testing (karyotype, chromosome microarray, and targeted DNA testing when indicated). Creatine kinase and TSH may be considered when motor concerns are present. An EEG is recommended when seizures are suspected or when there is language regression. Brain imaging is recommended if macrocephaly, microcephaly, or abnormal tone/motor findings are present.
Treatment emphasizes early and structured intervention. Nonpharmacologic therapy includes behavioral programs at home and school, applied behavioral analysis (ABA) approaches (such as Discrete Trial Training), and development-focused ABA models (such as Pivotal Response Training, Floortime, and the Early Start Denver Model). Specialized educational approaches focusing on communication and life skills (such as TEACCH) are often helpful. Cognitive-behavioral therapy can reduce anxiety in higher-functioning individuals. Family and teacher education and highly structured environments support skill-building and daily functioning.
Medication does not “cure” ASD, but can reduce specific target symptoms. Risperidone and aripiprazole are FDA-approved for managing irritability associated with ASD. Other medications are used off-label depending on symptoms, including SSRIs or atypical antipsychotics for obsessive or ritualistic behaviors, atypical antipsychotics and other agents for aggression or self-injury, stimulants or alpha-2 agonists for hyperactivity/inattention, and SSRIs or other agents for anxiety or depression. Catatonia is treated with lorazepam and sometimes electroconvulsive therapy, and antipsychotics should be avoided in catatonia because they can worsen it. Complementary approaches have limited evidence overall, though melatonin has preliminary evidence for sleep difficulties.
Most individuals with ASD require some level of support as adults. DSM-5-TR describes severity levels based on support needs, from Level 1 (requiring support) to Level 3 (requiring very substantial support). Better outcomes are linked to early identification and intervention, development of functional spoken language, and capacity for inclusion with typical peers. Poorer outcomes are associated with lack of joint attention by age 4, absence of functional speech by age 5, intellectual disability, seizures, significant comorbid medical or psychiatric conditions, and pervasive social disengagement. Referrals may involve specialists for diagnosis (developmental pediatrics, child psychiatry, psychology, neurology, genetics) and therapy services (speech-language therapy, occupational therapy, behavioral therapy), as well as support for caregivers and school planning.
There is no scientific evidence linking childhood vaccination to the development of ASD. Many individuals with ASD have increased rates of medical issues such as sleep problems, gastrointestinal concerns, and oral health problems, and psychiatric comorbidities are common across the lifespan.

- Published on
Emergency and Acute Medicine – Lithium Poisoning
Basic description
Lithium is rapidly absorbed from the gastrointestinal tract, with peak serum levels occurring at 2–4 hours for regular-release formulations and 4–12 hours for sustained-release formulations. Distribution into tissues is slow, requiring at least 6 hours, and toxicity correlates with intracellular rather than serum concentrations. Lithium is not metabolized and is excreted unchanged by the kidneys, where it is reabsorbed in the proximal tubule via sodium transport mechanisms. It has a narrow therapeutic window, with therapeutic levels of 0.6–1.2 mEq/L measured after distribution. Elimination half-life is approximately 20–24 hours but is prolonged in chronic users.
Etiology
Toxicity occurs due to overdose or conditions that reduce renal clearance. Acute risk factors include dehydration and overdose, while chronic risk factors include advanced age, renal failure, congestive heart failure, hypertension, diabetes mellitus, low-salt diets, dose changes, and drug interactions. Medications that increase lithium levels include NSAIDs, thiazide diuretics, ACE inhibitors, phenytoin, tricyclic antidepressants, and phenothiazines.
Diagnosis: signs and symptoms
Acute toxicity is often less severe initially and dominated by gastrointestinal symptoms such as nausea, vomiting, diarrhea, and abdominal pain. Neurologic findings progress from fine tremor and weakness to ataxia, slurred speech, blurred vision, coarse tremor, hyperreflexia, confusion, seizures, coma, clonus, and extrapyramidal symptoms. Chronic toxicity more commonly produces neurologic manifestations, including parkinsonism, psychosis, and persistent cognitive deficits. Additional findings include nephrogenic diabetes insipidus, interstitial nephritis, hypothyroidism, leukocytosis, and dermatologic changes. Cardiac effects include QT prolongation, ST depression, T-wave flattening, and rarely serious dysrhythmias.
Essential workup
Serum lithium levels must be interpreted based on timing and type of exposure. Because distribution is delayed, levels should be repeated every 2 hours to assess trends. Acute toxicity may show high levels with minimal symptoms early, whereas chronic toxicity correlates more closely with symptoms at lower levels. Toxicity is generally associated with levels >4 mEq/L in acute ingestion, >3 mEq/L in acute-on-chronic ingestion, and >1.5 mEq/L in chronic toxicity.
Diagnosis tests and interpretation
Laboratory evaluation includes electrolytes, BUN, creatinine, glucose, and urinalysis to assess renal function and hydration status. Aspirin and acetaminophen levels should be checked if coingestion is suspected. ECG monitoring is indicated to detect conduction abnormalities.
Differential diagnosis
Consider lithium toxicity in patients with altered mental status and neuromuscular findings. Other considerations include hypoglycemia, cholinergic poisoning, heavy-metal toxicity, neuroleptic overdose, envenomation, and strychnine poisoning.
Treatment and stabilization
Initial management follows ABC principles with IV access, cardiac monitoring, and correction of hypoglycemia if present. Benzodiazepines are used for seizure control. Gastrointestinal decontamination is limited; activated charcoal is ineffective for lithium but may be given if coingestants are suspected. Whole-bowel irrigation with polyethylene glycol is recommended for sustained-release preparations if no contraindications exist. Aggressive isotonic saline hydration is the cornerstone of therapy, enhancing renal clearance and reducing tubular reabsorption. Diuretics and sodium bicarbonate are not recommended.
Enhanced elimination
Hemodialysis is indicated in severe cases, including progressive neurologic symptoms, renal insufficiency, significant altered mental status, ventricular dysrhythmias, or very high lithium levels. Suggested thresholds include >4–5 mEq/L in acute ingestion and >2.5–3 mEq/L in chronic toxicity. Rebound lithium levels due to redistribution are common, and repeat dialysis may be required until levels fall below 1 mEq/L.
Disposition and follow-up
Admission is required for symptomatic patients, those requiring hemodialysis, intentional ingestions, or persistently elevated lithium levels despite treatment. ICU admission is indicated for severe neurologic toxicity. Asymptomatic patients with decreasing levels and serum lithium <2 mEq/L may be discharged with close follow-up if ingestion was unintentional. Psychiatric consultation is required for intentional overdose, and outpatient follow-up should ensure appropriate dosing and monitoring.
Key points
Serum lithium levels must be interpreted in clinical context and timing of exposure. Hydration is the most effective initial therapy. Early recognition of chronic toxicity and timely use of hemodialysis can prevent permanent neurologic sequelae.
Basic description
Lithium is rapidly absorbed from the gastrointestinal tract, with peak serum levels occurring at 2–4 hours for regular-release formulations and 4–12 hours for sustained-release formulations. Distribution into tissues is slow, requiring at least 6 hours, and toxicity correlates with intracellular rather than serum concentrations. Lithium is not metabolized and is excreted unchanged by the kidneys, where it is reabsorbed in the proximal tubule via sodium transport mechanisms. It has a narrow therapeutic window, with therapeutic levels of 0.6–1.2 mEq/L measured after distribution. Elimination half-life is approximately 20–24 hours but is prolonged in chronic users.
Etiology
Toxicity occurs due to overdose or conditions that reduce renal clearance. Acute risk factors include dehydration and overdose, while chronic risk factors include advanced age, renal failure, congestive heart failure, hypertension, diabetes mellitus, low-salt diets, dose changes, and drug interactions. Medications that increase lithium levels include NSAIDs, thiazide diuretics, ACE inhibitors, phenytoin, tricyclic antidepressants, and phenothiazines.
Diagnosis: signs and symptoms
Acute toxicity is often less severe initially and dominated by gastrointestinal symptoms such as nausea, vomiting, diarrhea, and abdominal pain. Neurologic findings progress from fine tremor and weakness to ataxia, slurred speech, blurred vision, coarse tremor, hyperreflexia, confusion, seizures, coma, clonus, and extrapyramidal symptoms. Chronic toxicity more commonly produces neurologic manifestations, including parkinsonism, psychosis, and persistent cognitive deficits. Additional findings include nephrogenic diabetes insipidus, interstitial nephritis, hypothyroidism, leukocytosis, and dermatologic changes. Cardiac effects include QT prolongation, ST depression, T-wave flattening, and rarely serious dysrhythmias.
Essential workup
Serum lithium levels must be interpreted based on timing and type of exposure. Because distribution is delayed, levels should be repeated every 2 hours to assess trends. Acute toxicity may show high levels with minimal symptoms early, whereas chronic toxicity correlates more closely with symptoms at lower levels. Toxicity is generally associated with levels >4 mEq/L in acute ingestion, >3 mEq/L in acute-on-chronic ingestion, and >1.5 mEq/L in chronic toxicity.
Diagnosis tests and interpretation
Laboratory evaluation includes electrolytes, BUN, creatinine, glucose, and urinalysis to assess renal function and hydration status. Aspirin and acetaminophen levels should be checked if coingestion is suspected. ECG monitoring is indicated to detect conduction abnormalities.
Differential diagnosis
Consider lithium toxicity in patients with altered mental status and neuromuscular findings. Other considerations include hypoglycemia, cholinergic poisoning, heavy-metal toxicity, neuroleptic overdose, envenomation, and strychnine poisoning.
Treatment and stabilization
Initial management follows ABC principles with IV access, cardiac monitoring, and correction of hypoglycemia if present. Benzodiazepines are used for seizure control. Gastrointestinal decontamination is limited; activated charcoal is ineffective for lithium but may be given if coingestants are suspected. Whole-bowel irrigation with polyethylene glycol is recommended for sustained-release preparations if no contraindications exist. Aggressive isotonic saline hydration is the cornerstone of therapy, enhancing renal clearance and reducing tubular reabsorption. Diuretics and sodium bicarbonate are not recommended.
Enhanced elimination
Hemodialysis is indicated in severe cases, including progressive neurologic symptoms, renal insufficiency, significant altered mental status, ventricular dysrhythmias, or very high lithium levels. Suggested thresholds include >4–5 mEq/L in acute ingestion and >2.5–3 mEq/L in chronic toxicity. Rebound lithium levels due to redistribution are common, and repeat dialysis may be required until levels fall below 1 mEq/L.
Disposition and follow-up
Admission is required for symptomatic patients, those requiring hemodialysis, intentional ingestions, or persistently elevated lithium levels despite treatment. ICU admission is indicated for severe neurologic toxicity. Asymptomatic patients with decreasing levels and serum lithium <2 mEq/L may be discharged with close follow-up if ingestion was unintentional. Psychiatric consultation is required for intentional overdose, and outpatient follow-up should ensure appropriate dosing and monitoring.
Key points
Serum lithium levels must be interpreted in clinical context and timing of exposure. Hydration is the most effective initial therapy. Early recognition of chronic toxicity and timely use of hemodialysis can prevent permanent neurologic sequelae.
- Published on
Emergency and Acute Medicine – Lyme Disease
Basics description
Lyme disease is the most common tick-borne illness in North America. It is endemic in the northeastern United States, the upper Midwest, and parts of northwestern California. The disease follows a multisystem course and may present with dermatologic, neurologic, cardiac, and musculoskeletal manifestations.
Etiology and pathophysiology
Lyme disease is caused by the spirochete Borrelia burgdorferi, which is transmitted through the bite of an Ixodes tick, most commonly Ixodes dammini (the deer tick). Transmission occurs primarily between April and November, with 80–90% of cases in the summer months. Fewer than half of patients recall a tick bite. Disease pathogenesis involves organism-induced local inflammation, cytokine release, and autoimmune mechanisms. There is no person-to-person transmission. A related spirochete, Borrelia miyamotoi, has also been identified as a cause of a Lyme-like illness.
Clinical presentation and stages
Lyme disease progresses through three clinical stages, although not all patients experience every stage.
Stage I, or early localized disease, begins days to weeks after a tick bite. The hallmark finding is erythema chronicum migrans, a pathognomonic expanding annular rash greater than 5 cm in diameter, often with central clearing and a red outer border (“bull’s-eye” rash). Associated symptoms include regional lymphadenopathy, low-grade fever, headache, myalgias, arthralgias, fatigue, and malaise.
Stage II, or early disseminated disease, occurs days to weeks after infection and is characterized by intermittent, fluctuating symptoms. Neurologic involvement includes the triad of aseptic meningitis, cranial neuritis, and radiculoneuritis, with facial (Bell) palsy being the most common cranial nerve deficit. Cardiac manifestations may include tachycardia, bradycardia, atrioventricular block, and myopericarditis. Rash may be absent at this stage. Prognosis is generally good with appropriate treatment.
Stage III, or late disease, develops months to years after initial infection. Musculoskeletal involvement is most common, typically presenting as recurrent monoarticular or oligoarticular arthritis, most often affecting the knee. Dermatologic findings may include acrodermatitis chronica atrophicans on the extensor surfaces of the extremities. Less commonly, late neurologic complications such as chronic polyneuropathy or encephalopathy may occur. A Jarisch–Herxheimer reaction, consisting of transient symptom worsening shortly after treatment initiation, may be seen.
Special populations
Children are more likely than adults to present with fever and may lack a history of erythema migrans. Facial palsy in pediatric patients is frequently associated with aseptic meningitis. Cardiac involvement may be asymptomatic but detectable on ECG. With appropriate treatment, children generally have excellent long-term outcomes. There is no clear evidence that Lyme disease during pregnancy causes fetal harm.
Diagnosis and evaluation
Lyme disease is primarily a clinical diagnosis. The presence of erythema migrans eliminates the need for serologic testing. In patients without the classic rash, serologic testing with ELISA followed by confirmatory Western blot is indicated. Lumbar puncture is reserved for patients with meningeal signs, arthrocentesis for acute arthritis, and ECG for suspected cardiac involvement.
Laboratory findings are nonspecific and may include elevated erythrocyte sedimentation rate, mild cytopenias, or elevated liver enzymes. Cerebrospinal fluid analysis may show pleocytosis and elevated protein in neuroborreliosis.
Differential diagnosis
The differential diagnosis includes other tick-borne illnesses, viral meningitis, septic arthritis, rheumatic fever, syphilis, parvovirus B19 infection, infectious endocarditis, juvenile idiopathic arthritis, fibromyalgia, and chronic fatigue syndrome.
Treatment
Initial stabilization focuses on hydration, IV access for neurologic or cardiac involvement, and cardiac monitoring when indicated. Tick removal should be performed promptly using blunt forceps applied close to the skin.
Antibiotic therapy depends on disease stage and severity. Early localized disease is treated with oral antibiotics such as amoxicillin, doxycycline (for patients ≥8 years old and nonpregnant), or cefuroxime. Disseminated disease with mild neurologic involvement may be treated orally, while meningitis, significant carditis, or severe arthritis requires parenteral therapy, typically with ceftriaxone, cefotaxime, or penicillin G. Late disease usually requires prolonged parenteral therapy. NSAIDs are used for arthralgias and arthritis, and aspirin may be used adjunctively in cardiac involvement.
Disposition and follow-up
Hospital admission is required for patients with meningoencephalitis or significant cardiac involvement requiring telemetry or ICU monitoring. Patients with uncomplicated disease treated with oral antibiotics may be discharged with close outpatient follow-up.
Key points
Early recognition and treatment of Lyme disease prevent late complications. The presence of erythema migrans is diagnostic and should prompt immediate treatment without waiting for serology. Clinicians should remain vigilant for coinfections such as anaplasmosis and babesiosis in endemic regions, and prolonged treatment is required for later-stage organ involvement.
Basics description
Lyme disease is the most common tick-borne illness in North America. It is endemic in the northeastern United States, the upper Midwest, and parts of northwestern California. The disease follows a multisystem course and may present with dermatologic, neurologic, cardiac, and musculoskeletal manifestations.
Etiology and pathophysiology
Lyme disease is caused by the spirochete Borrelia burgdorferi, which is transmitted through the bite of an Ixodes tick, most commonly Ixodes dammini (the deer tick). Transmission occurs primarily between April and November, with 80–90% of cases in the summer months. Fewer than half of patients recall a tick bite. Disease pathogenesis involves organism-induced local inflammation, cytokine release, and autoimmune mechanisms. There is no person-to-person transmission. A related spirochete, Borrelia miyamotoi, has also been identified as a cause of a Lyme-like illness.
Clinical presentation and stages
Lyme disease progresses through three clinical stages, although not all patients experience every stage.
Stage I, or early localized disease, begins days to weeks after a tick bite. The hallmark finding is erythema chronicum migrans, a pathognomonic expanding annular rash greater than 5 cm in diameter, often with central clearing and a red outer border (“bull’s-eye” rash). Associated symptoms include regional lymphadenopathy, low-grade fever, headache, myalgias, arthralgias, fatigue, and malaise.
Stage II, or early disseminated disease, occurs days to weeks after infection and is characterized by intermittent, fluctuating symptoms. Neurologic involvement includes the triad of aseptic meningitis, cranial neuritis, and radiculoneuritis, with facial (Bell) palsy being the most common cranial nerve deficit. Cardiac manifestations may include tachycardia, bradycardia, atrioventricular block, and myopericarditis. Rash may be absent at this stage. Prognosis is generally good with appropriate treatment.
Stage III, or late disease, develops months to years after initial infection. Musculoskeletal involvement is most common, typically presenting as recurrent monoarticular or oligoarticular arthritis, most often affecting the knee. Dermatologic findings may include acrodermatitis chronica atrophicans on the extensor surfaces of the extremities. Less commonly, late neurologic complications such as chronic polyneuropathy or encephalopathy may occur. A Jarisch–Herxheimer reaction, consisting of transient symptom worsening shortly after treatment initiation, may be seen.
Special populations
Children are more likely than adults to present with fever and may lack a history of erythema migrans. Facial palsy in pediatric patients is frequently associated with aseptic meningitis. Cardiac involvement may be asymptomatic but detectable on ECG. With appropriate treatment, children generally have excellent long-term outcomes. There is no clear evidence that Lyme disease during pregnancy causes fetal harm.
Diagnosis and evaluation
Lyme disease is primarily a clinical diagnosis. The presence of erythema migrans eliminates the need for serologic testing. In patients without the classic rash, serologic testing with ELISA followed by confirmatory Western blot is indicated. Lumbar puncture is reserved for patients with meningeal signs, arthrocentesis for acute arthritis, and ECG for suspected cardiac involvement.
Laboratory findings are nonspecific and may include elevated erythrocyte sedimentation rate, mild cytopenias, or elevated liver enzymes. Cerebrospinal fluid analysis may show pleocytosis and elevated protein in neuroborreliosis.
Differential diagnosis
The differential diagnosis includes other tick-borne illnesses, viral meningitis, septic arthritis, rheumatic fever, syphilis, parvovirus B19 infection, infectious endocarditis, juvenile idiopathic arthritis, fibromyalgia, and chronic fatigue syndrome.
Treatment
Initial stabilization focuses on hydration, IV access for neurologic or cardiac involvement, and cardiac monitoring when indicated. Tick removal should be performed promptly using blunt forceps applied close to the skin.
Antibiotic therapy depends on disease stage and severity. Early localized disease is treated with oral antibiotics such as amoxicillin, doxycycline (for patients ≥8 years old and nonpregnant), or cefuroxime. Disseminated disease with mild neurologic involvement may be treated orally, while meningitis, significant carditis, or severe arthritis requires parenteral therapy, typically with ceftriaxone, cefotaxime, or penicillin G. Late disease usually requires prolonged parenteral therapy. NSAIDs are used for arthralgias and arthritis, and aspirin may be used adjunctively in cardiac involvement.
Disposition and follow-up
Hospital admission is required for patients with meningoencephalitis or significant cardiac involvement requiring telemetry or ICU monitoring. Patients with uncomplicated disease treated with oral antibiotics may be discharged with close outpatient follow-up.
Key points
Early recognition and treatment of Lyme disease prevent late complications. The presence of erythema migrans is diagnostic and should prompt immediate treatment without waiting for serology. Clinicians should remain vigilant for coinfections such as anaplasmosis and babesiosis in endemic regions, and prolonged treatment is required for later-stage organ involvement.