- Published on
Emergency and Acute Medicine – Mallory–Weiss Syndrome
Basics description
Mallory–Weiss syndrome is a partial-thickness, longitudinal mucosal tear of the distal esophagus or proximal stomach. It results from a sudden increase in intra-abdominal or transgastric pressure, leading to mild to moderate submucosal arterial or venous bleeding. Endoscopic observations suggest that transient “mushrooming” of the stomach into the esophagus during forceful retching contributes to tear formation. It accounts for approximately 5% of all cases of upper gastrointestinal bleeding.
Etiology and risk factors
The condition is most commonly associated with forceful vomiting or retching but may also occur with coughing, laughing, lifting, straining, blunt abdominal trauma, seizures, childbirth, or cardiopulmonary resuscitation. Alcohol use, particularly following binge drinking, and the presence of a hiatal hernia are significant risk factors. More severe bleeding is seen in patients with portal hypertension, esophageal varices, or underlying coagulopathy.
Diagnosis signs and symptoms
Patients typically report multiple episodes of nonbloody vomiting or retching followed by hematemesis. Most bleeding is self-limited, though severe or life-threatening hemorrhage can occur. Associated symptoms include epigastric or back pain and signs of volume depletion such as dizziness, light-headedness, or syncope. Physical examination may reveal hematemesis, melena, postural hypotension, or shock in severe cases.
Essential workup and diagnostic testing
Initial evaluation includes a complete blood count and rectal examination for occult blood. Additional laboratory studies include coagulation parameters, electrolytes, renal function tests, liver function tests, and type and cross-match if bleeding is significant. Upright chest radiography may be obtained to exclude esophageal or gastric perforation. Upper endoscopy is the diagnostic and therapeutic modality of choice, allowing direct visualization and management of the bleeding source.
Differential diagnosis
Conditions to consider include nasopharyngeal bleeding, hemoptysis, esophageal rupture (Boerhaave syndrome), esophagitis, gastritis, peptic ulcer disease, variceal bleeding, malignancy, and vascular-enteric fistula.
Treatment and emergency management
Initial management focuses on airway protection, oxygenation, and hemodynamic stabilization with large-bore intravenous access and fluid resuscitation. Blood transfusion is indicated for ongoing bleeding or hemodynamic instability. Most cases resolve with conservative therapy. Persistent or active bleeding requires urgent endoscopic intervention, which may include injection therapy, thermal coagulation, clipping, or band ligation. Proton pump inhibitors and antiemetics are used adjunctively. Surgical or angiographic intervention is reserved for refractory cases.
Disposition and follow-up
Patients with ongoing or massive hemorrhage, hemodynamic instability, significant comorbidities, or advanced age require ICU admission. Stable patients with minimal bleeding that has resolved and stable hematocrit may be admitted for observation or, in select cases, discharged with close follow-up and outpatient endoscopy.
Key points and cautions
Early placement of large-bore intravenous access and prompt resuscitation are critical in upper gastrointestinal bleeding. Early gastroenterology consultation is essential when bleeding is significant. Rebleeding most often occurs within the first 24 hours and is more likely in patients with coagulopathy or portal hypertension.
Basics description
Mallory–Weiss syndrome is a partial-thickness, longitudinal mucosal tear of the distal esophagus or proximal stomach. It results from a sudden increase in intra-abdominal or transgastric pressure, leading to mild to moderate submucosal arterial or venous bleeding. Endoscopic observations suggest that transient “mushrooming” of the stomach into the esophagus during forceful retching contributes to tear formation. It accounts for approximately 5% of all cases of upper gastrointestinal bleeding.
Etiology and risk factors
The condition is most commonly associated with forceful vomiting or retching but may also occur with coughing, laughing, lifting, straining, blunt abdominal trauma, seizures, childbirth, or cardiopulmonary resuscitation. Alcohol use, particularly following binge drinking, and the presence of a hiatal hernia are significant risk factors. More severe bleeding is seen in patients with portal hypertension, esophageal varices, or underlying coagulopathy.
Diagnosis signs and symptoms
Patients typically report multiple episodes of nonbloody vomiting or retching followed by hematemesis. Most bleeding is self-limited, though severe or life-threatening hemorrhage can occur. Associated symptoms include epigastric or back pain and signs of volume depletion such as dizziness, light-headedness, or syncope. Physical examination may reveal hematemesis, melena, postural hypotension, or shock in severe cases.
Essential workup and diagnostic testing
Initial evaluation includes a complete blood count and rectal examination for occult blood. Additional laboratory studies include coagulation parameters, electrolytes, renal function tests, liver function tests, and type and cross-match if bleeding is significant. Upright chest radiography may be obtained to exclude esophageal or gastric perforation. Upper endoscopy is the diagnostic and therapeutic modality of choice, allowing direct visualization and management of the bleeding source.
Differential diagnosis
Conditions to consider include nasopharyngeal bleeding, hemoptysis, esophageal rupture (Boerhaave syndrome), esophagitis, gastritis, peptic ulcer disease, variceal bleeding, malignancy, and vascular-enteric fistula.
Treatment and emergency management
Initial management focuses on airway protection, oxygenation, and hemodynamic stabilization with large-bore intravenous access and fluid resuscitation. Blood transfusion is indicated for ongoing bleeding or hemodynamic instability. Most cases resolve with conservative therapy. Persistent or active bleeding requires urgent endoscopic intervention, which may include injection therapy, thermal coagulation, clipping, or band ligation. Proton pump inhibitors and antiemetics are used adjunctively. Surgical or angiographic intervention is reserved for refractory cases.
Disposition and follow-up
Patients with ongoing or massive hemorrhage, hemodynamic instability, significant comorbidities, or advanced age require ICU admission. Stable patients with minimal bleeding that has resolved and stable hematocrit may be admitted for observation or, in select cases, discharged with close follow-up and outpatient endoscopy.
Key points and cautions
Early placement of large-bore intravenous access and prompt resuscitation are critical in upper gastrointestinal bleeding. Early gastroenterology consultation is essential when bleeding is significant. Rebleeding most often occurs within the first 24 hours and is more likely in patients with coagulopathy or portal hypertension.
- Published on
Emergency and Acute Medicine – Malaria
Basics description
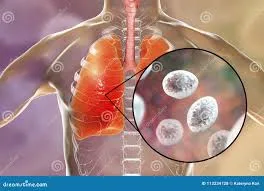
Malaria is a protozoan infection transmitted through the bite of an infected female Anopheles mosquito. The incubation period is typically 8–16 days, although symptoms may be delayed for months depending on the species. The periodic nature of malaria symptoms reflects the parasite life cycle, which includes an exoerythrocytic phase in the liver followed by an erythrocytic phase in red blood cells. Fever corresponds to synchronous red blood cell lysis and release of merozoites into the circulation.
Pathophysiology and species differences
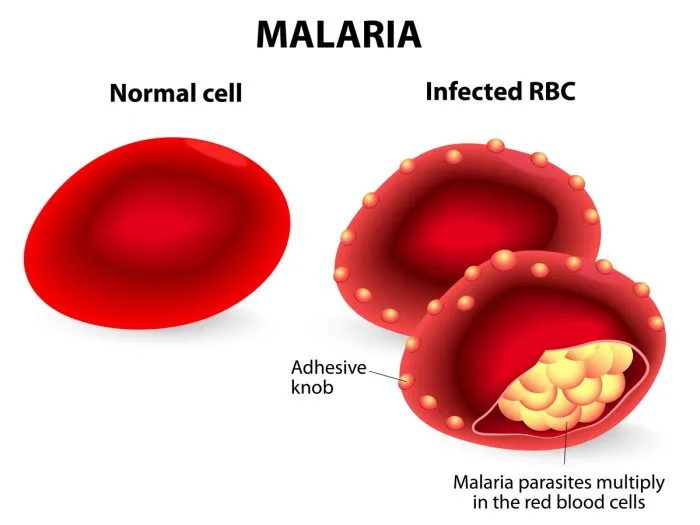
In the exoerythrocytic phase, sporozoites migrate to the liver and multiply into merozoites. These then enter the bloodstream and invade red blood cells, where replication leads to cell rupture every 48–72 hours. Plasmodium falciparum causes most severe disease and nearly all malaria-related deaths. It infects red blood cells of all ages, leading to profound hemolysis, anemia, and capillary obstruction with resultant end-organ hypoxia. Plasmodium vivax and Plasmodium ovale may cause relapsing disease due to dormant liver forms (hypnozoites), while Plasmodium malariae can persist at low levels in the bloodstream for decades.
Etiology and transmission
Transmission usually occurs via mosquito bite but may also result from blood transfusions or shared needles. Rare autochthonous transmission has been reported in North America due to the presence of Anopheles mosquitoes. Post-traumatic immunosuppression may precipitate relapse in individuals previously exposed in endemic regions. Children with sickle cell trait have partial protection, while pregnant patients—especially primigravidas—are at higher risk for severe disease.
Diagnosis signs and symptoms
Most patients become symptomatic within one year of exposure. Common symptoms include malaise, chills, fever greater than 38°C, myalgias, arthralgias, and orthostatic hypotension. The classic malaria paroxysm consists of chills followed by high fever and subsequent diaphoresis, although this pattern is uncommon in clinical practice. Hemolysis may lead to jaundice, dark urine (blackwater fever), anemia, and splenomegaly. Severe manifestations include cerebral malaria with headache, altered mental status, seizures, or coma; pulmonary edema; renal failure; abnormal bleeding; and circulatory collapse.
Essential workup and diagnostic testing
Diagnosis relies on oil immersion light microscopy of thick and thin Giemsa-stained blood smears, which demonstrate intraerythrocytic parasites. At least three negative smears over 48 hours are required to exclude malaria. Laboratory findings commonly include anemia, thrombocytopenia, leukocytopenia, elevated bilirubin and lactate dehydrogenase, electrolyte abnormalities, and evidence of renal or hepatic dysfunction. Chest radiography may reveal pulmonary edema. Advanced testing such as antigen detection assays or PCR can identify Plasmodium species and mixed infections. Lumbar puncture may be necessary to distinguish cerebral malaria from meningitis.
Differential diagnosis
The differential includes meningitis, encephalitis, sepsis, acute renal failure, viral hepatitis, acute hemolytic anemia, hypoglycemic coma, and heat stroke. In travelers or immigrants from endemic areas, malaria must be considered in any febrile illness.
Treatment and emergency management
Initial stabilization includes airway, breathing, and circulation management, isotonic fluid resuscitation for hypotension, active cooling for hyperthermia, and treatment of hypoglycemia or seizures as indicated. Antimalarial therapy depends on species, severity, and geographic resistance patterns, with artemisinin-based combination therapies recommended as first-line treatment worldwide. Severe P. falciparum infection requires intravenous artesunate. Supportive care for complications such as anemia, renal failure, and cerebral edema is critical.
Disposition and follow-up
ICU admission is indicated for severe P. falciparum malaria, high parasitemia, neurologic involvement, or inability to tolerate oral therapy. Patients with non–P. falciparum malaria who are clinically stable and able to take oral medications may be treated as outpatients with close follow-up.
Pearls and pitfalls
Malaria should always be considered in patients with fever and compatible exposure history, even months after travel. Fever patterns are unreliable, and early recognition is essential, as delays in treatment—especially with P. falciparum—are associated with high mortality.
Basics description
Malaria is a protozoan infection transmitted through the bite of an infected female Anopheles mosquito. The incubation period is typically 8–16 days, although symptoms may be delayed for months depending on the species. The periodic nature of malaria symptoms reflects the parasite life cycle, which includes an exoerythrocytic phase in the liver followed by an erythrocytic phase in red blood cells. Fever corresponds to synchronous red blood cell lysis and release of merozoites into the circulation.
Pathophysiology and species differences
In the exoerythrocytic phase, sporozoites migrate to the liver and multiply into merozoites. These then enter the bloodstream and invade red blood cells, where replication leads to cell rupture every 48–72 hours. Plasmodium falciparum causes most severe disease and nearly all malaria-related deaths. It infects red blood cells of all ages, leading to profound hemolysis, anemia, and capillary obstruction with resultant end-organ hypoxia. Plasmodium vivax and Plasmodium ovale may cause relapsing disease due to dormant liver forms (hypnozoites), while Plasmodium malariae can persist at low levels in the bloodstream for decades.
Etiology and transmission
Transmission usually occurs via mosquito bite but may also result from blood transfusions or shared needles. Rare autochthonous transmission has been reported in North America due to the presence of Anopheles mosquitoes. Post-traumatic immunosuppression may precipitate relapse in individuals previously exposed in endemic regions. Children with sickle cell trait have partial protection, while pregnant patients—especially primigravidas—are at higher risk for severe disease.
Diagnosis signs and symptoms
Most patients become symptomatic within one year of exposure. Common symptoms include malaise, chills, fever greater than 38°C, myalgias, arthralgias, and orthostatic hypotension. The classic malaria paroxysm consists of chills followed by high fever and subsequent diaphoresis, although this pattern is uncommon in clinical practice. Hemolysis may lead to jaundice, dark urine (blackwater fever), anemia, and splenomegaly. Severe manifestations include cerebral malaria with headache, altered mental status, seizures, or coma; pulmonary edema; renal failure; abnormal bleeding; and circulatory collapse.
Essential workup and diagnostic testing
Diagnosis relies on oil immersion light microscopy of thick and thin Giemsa-stained blood smears, which demonstrate intraerythrocytic parasites. At least three negative smears over 48 hours are required to exclude malaria. Laboratory findings commonly include anemia, thrombocytopenia, leukocytopenia, elevated bilirubin and lactate dehydrogenase, electrolyte abnormalities, and evidence of renal or hepatic dysfunction. Chest radiography may reveal pulmonary edema. Advanced testing such as antigen detection assays or PCR can identify Plasmodium species and mixed infections. Lumbar puncture may be necessary to distinguish cerebral malaria from meningitis.
Differential diagnosis
The differential includes meningitis, encephalitis, sepsis, acute renal failure, viral hepatitis, acute hemolytic anemia, hypoglycemic coma, and heat stroke. In travelers or immigrants from endemic areas, malaria must be considered in any febrile illness.
Treatment and emergency management
Initial stabilization includes airway, breathing, and circulation management, isotonic fluid resuscitation for hypotension, active cooling for hyperthermia, and treatment of hypoglycemia or seizures as indicated. Antimalarial therapy depends on species, severity, and geographic resistance patterns, with artemisinin-based combination therapies recommended as first-line treatment worldwide. Severe P. falciparum infection requires intravenous artesunate. Supportive care for complications such as anemia, renal failure, and cerebral edema is critical.
Disposition and follow-up
ICU admission is indicated for severe P. falciparum malaria, high parasitemia, neurologic involvement, or inability to tolerate oral therapy. Patients with non–P. falciparum malaria who are clinically stable and able to take oral medications may be treated as outpatients with close follow-up.
Pearls and pitfalls
Malaria should always be considered in patients with fever and compatible exposure history, even months after travel. Fever patterns are unreliable, and early recognition is essential, as delays in treatment—especially with P. falciparum—are associated with high mortality.
- Published on
Emergency and Acute Medicine – Lymphogranuloma Venereum
Basics description
Lymphogranuloma venereum (LGV) is a sexually transmitted infection characterized by an initial painless genital lesion followed by regional lymphatic spread. The disease progresses through primary, secondary, and tertiary stages and is responsive to appropriate antibacterial therapy in early phases. LGV is endemic in Southeast Asia, Latin America, parts of Africa, and the Caribbean, with increasing incidence among men who have sex with men. It is also known as struma, tropical bubo, or Nicolas–Favre–Durand disease.
Etiology
LGV is caused by Chlamydia trachomatis serotypes L1, L2, and L3.
Diagnosis signs and symptoms
The primary stage occurs after an incubation period of 3–30 days and presents as a painless papule, pustule, vesicle, or ulcer in the anogenital region. These lesions are transient, often last only a few days, and are frequently unnoticed. The secondary stage develops 1–3 weeks later with systemic symptoms such as fever, malaise, and myalgias, along with tender inguinal lymphadenopathy that may be unilateral or bilateral. Large, fluctuant lymph nodes (buboes) can ulcerate and drain purulent material. Proctitis is common in anal-receptive patients and may cause rectal bleeding, tenesmus, and constipation. The tertiary stage occurs in untreated disease and results in chronic inflammatory changes including proctocolitis, strictures, fistulae, and elephantiasis of the genitalia or lower extremities, mimicking inflammatory bowel disease.
Physical examination
Findings in the primary stage include a painless anogenital lesion. During the secondary stage, tender inguinal or femoral lymphadenopathy is typical, with buboes forming in up to two-thirds of cases. The “groove sign,” caused by lymph node enlargement above and below the inguinal ligament, may be seen. Anal-receptive patients may demonstrate signs of hemorrhagic proctocolitis. Advanced tertiary disease shows chronic inflammatory damage with strictures, fistulae, and elephantiasis.
Diagnosis tests and interpretation
Routine chlamydia nucleic acid amplification tests do not differentiate LGV strains. Diagnosis is based on clinical suspicion, epidemiologic context, and serologic testing. Complement fixation titers greater than 1:64 support the diagnosis. False-positive VDRL tests may occur. Bubo aspiration is specific but rarely practical and is not routinely required.
Differential diagnosis
Conditions to consider include genital herpes, syphilis, chancroid, and granuloma inguinale. Compared with LGV, syphilis typically causes nontender lymphadenopathy with a longer incubation period, while chancroid presents with painful ulcers and granuloma inguinale causes painless, friable lesions that bleed easily.
Treatment
No prehospital or emergency stabilization is typically required. Large, fluctuant buboes may require aspiration or drainage to reduce pain and scarring. First-line therapy is doxycycline 100 mg orally twice daily for 21 days. Alternative regimens include azithromycin 1 g orally weekly for 3 weeks or erythromycin 500 mg orally four times daily for 3 weeks. In pregnancy or during lactation, erythromycin is the recommended treatment.
Disposition and follow-up
Hospitalization is rarely necessary and is reserved for patients with severe systemic illness or complications. Most immunocompetent patients without systemic involvement can be discharged with outpatient management. Sexual partners within the previous 60 days should be tested and treated with appropriate antichlamydial therapy. Follow-up is essential to confirm cure, particularly in cases of rectal involvement, which may require retreatment.
Pearls and pitfalls
LGV should be suspected in at-risk populations, particularly men who have sex with men presenting with inguinal lymphadenopathy or proctitis. Early recognition and treatment are critical to prevent progression to tertiary disease, which may cause irreversible tissue damage and is less responsive to antibiotics alone.
Basics description
Lymphogranuloma venereum (LGV) is a sexually transmitted infection characterized by an initial painless genital lesion followed by regional lymphatic spread. The disease progresses through primary, secondary, and tertiary stages and is responsive to appropriate antibacterial therapy in early phases. LGV is endemic in Southeast Asia, Latin America, parts of Africa, and the Caribbean, with increasing incidence among men who have sex with men. It is also known as struma, tropical bubo, or Nicolas–Favre–Durand disease.
Etiology
LGV is caused by Chlamydia trachomatis serotypes L1, L2, and L3.
Diagnosis signs and symptoms
The primary stage occurs after an incubation period of 3–30 days and presents as a painless papule, pustule, vesicle, or ulcer in the anogenital region. These lesions are transient, often last only a few days, and are frequently unnoticed. The secondary stage develops 1–3 weeks later with systemic symptoms such as fever, malaise, and myalgias, along with tender inguinal lymphadenopathy that may be unilateral or bilateral. Large, fluctuant lymph nodes (buboes) can ulcerate and drain purulent material. Proctitis is common in anal-receptive patients and may cause rectal bleeding, tenesmus, and constipation. The tertiary stage occurs in untreated disease and results in chronic inflammatory changes including proctocolitis, strictures, fistulae, and elephantiasis of the genitalia or lower extremities, mimicking inflammatory bowel disease.
Physical examination
Findings in the primary stage include a painless anogenital lesion. During the secondary stage, tender inguinal or femoral lymphadenopathy is typical, with buboes forming in up to two-thirds of cases. The “groove sign,” caused by lymph node enlargement above and below the inguinal ligament, may be seen. Anal-receptive patients may demonstrate signs of hemorrhagic proctocolitis. Advanced tertiary disease shows chronic inflammatory damage with strictures, fistulae, and elephantiasis.
Diagnosis tests and interpretation
Routine chlamydia nucleic acid amplification tests do not differentiate LGV strains. Diagnosis is based on clinical suspicion, epidemiologic context, and serologic testing. Complement fixation titers greater than 1:64 support the diagnosis. False-positive VDRL tests may occur. Bubo aspiration is specific but rarely practical and is not routinely required.
Differential diagnosis
Conditions to consider include genital herpes, syphilis, chancroid, and granuloma inguinale. Compared with LGV, syphilis typically causes nontender lymphadenopathy with a longer incubation period, while chancroid presents with painful ulcers and granuloma inguinale causes painless, friable lesions that bleed easily.
Treatment
No prehospital or emergency stabilization is typically required. Large, fluctuant buboes may require aspiration or drainage to reduce pain and scarring. First-line therapy is doxycycline 100 mg orally twice daily for 21 days. Alternative regimens include azithromycin 1 g orally weekly for 3 weeks or erythromycin 500 mg orally four times daily for 3 weeks. In pregnancy or during lactation, erythromycin is the recommended treatment.
Disposition and follow-up
Hospitalization is rarely necessary and is reserved for patients with severe systemic illness or complications. Most immunocompetent patients without systemic involvement can be discharged with outpatient management. Sexual partners within the previous 60 days should be tested and treated with appropriate antichlamydial therapy. Follow-up is essential to confirm cure, particularly in cases of rectal involvement, which may require retreatment.
Pearls and pitfalls
LGV should be suspected in at-risk populations, particularly men who have sex with men presenting with inguinal lymphadenopathy or proctitis. Early recognition and treatment are critical to prevent progression to tertiary disease, which may cause irreversible tissue damage and is less responsive to antibiotics alone.
- Published on
KembaraXtra-Medicine – Meningococcemia
Meningococcemia is a serious bacterial illness caused by Neisseria meningitidis. It can present in several clinical forms, ranging from mild, self-limited disease to rapidly fatal overwhelming sepsis. Other manifestations include meningococcal meningitis, chronic or occult meningococcemia, septic arthritis, and less commonly localized infections. Transmission occurs through close contact with an infected person or an asymptomatic carrier, with intimate kissing and cigarette smoking recognized as independent risk factors.
The disease is caused by Neisseria meningitidis, a gram-negative diplococcus with multiple serotypes. Although many serotypes exist, most infections are caused by serogroups A, B, C, X, Y, and W135, with serogroup B being the most common in the United States. The organism colonizes the human nasopharynx, the only natural reservoir, and may invade the bloodstream after penetrating nasopharyngeal epithelial cells. Circulating bacteria are normally cleared by the spleen, but meningococci produce a potent endotoxin, lipooligosaccharide, which plays a central role in vascular injury, skin manifestations, adrenal hemorrhage, and circulatory collapse. Carrier rates increase with age, reaching up to 30–40% in young adults, while young children have lower immunity and higher infection risk. Disease incidence peaks in fall and spring and is higher in crowded living conditions such as military barracks and dormitories.
Clinical presentation varies widely. Mild meningococcemia is the most common form and is often preceded by an upper respiratory infection. Patients develop fever, chills, myalgias, arthralgias, and malaise, and the illness may resolve spontaneously over several days. However, this form can progress to meningitis or overwhelming sepsis. Overwhelming meningococcal sepsis occurs in approximately 10% of cases and carries a high mortality rate, with most deaths occurring within the first 48 hours. The onset is abrupt, and early symptoms may appear deceptively mild, including low-grade tachycardia, tachypnea, hypotension, fever, vomiting, headache, rash, and muscle tenderness. Infants may present with lethargy, poor feeding, or a bulging fontanel.
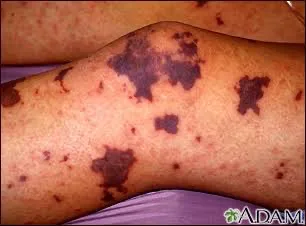
A characteristic rash is a key feature, often beginning as petechiae on the skin, mucous membranes, or conjunctivae, and progressing to purpura and ecchymoses. In severe cases, the rash may coalesce and become necrotic, leading to purpura fulminans. Rapid deterioration may follow, with hypotension, shock, metabolic acidosis, acute respiratory distress syndrome, and disseminated intravascular coagulation. Meningitis may or may not be present. A particularly severe complication is Waterhouse–Friderichsen syndrome, marked by bilateral adrenal hemorrhage, vasomotor collapse, and acute renal failure due to prolonged hypotension.
Chronic meningococcemia is uncommon and presents as a more indolent illness over weeks to months. Patients are often well appearing but experience recurrent fevers, chills, arthralgias, migratory polyarthritis, intermittent painful rashes on the extremities, and sometimes splenomegaly. Meningococcal meningitis presents with headache, fever, neck stiffness, altered mental status, lethargy, or obtundation. Septic arthritis may occur during active bacteremia and typically involves multiple joints with severe pain, swelling, effusion, and restricted movement. Less common manifestations include conjunctivitis, sinusitis, pneumonia, urethritis, salpingitis, prostatitis, myocarditis, and pericarditis.
Diagnosis is primarily clinical and should be suspected in any patient with fever, rash, and rapid clinical deterioration. Importantly, diagnostic evaluation must never delay resuscitation or antibiotic administration. Gram stain and cultures may be obtained from blood, cerebrospinal fluid, joint aspirates, sputum, urine, or scrapings of petechial or papular lesions, which may reveal intracellular or extracellular gram-negative diplococci. Laboratory findings may include leukocytosis early in disease, followed by leukopenia in severe cases, thrombocytopenia in association with purpura or DIC, metabolic acidosis, renal dysfunction, and abnormal coagulation parameters. Polymerase chain reaction testing is particularly useful when antibiotics have already been administered.
Management requires immediate stabilization with strict attention to airway, breathing, and circulation. Droplet precautions should be instituted, and public health authorities notified. Aggressive fluid resuscitation is essential for hypotension, with vasopressors such as dopamine, norepinephrine, or epinephrine if shock persists. Early endotracheal intubation is indicated for severe hypoxia, acidosis, or altered mental status. Empiric intravenous antibiotics must be started promptly, typically with a third-generation cephalosporin, and later tailored once the diagnosis is confirmed. Penicillin remains an option for proven susceptible infections, while chloramphenicol may be used in penicillin-allergic patients.
Supportive care includes management of metabolic acidosis, monitoring of urine output, treatment of DIC with blood products as indicated, and consideration of corticosteroids in select cases, particularly with adrenal involvement. Surgical intervention, including debridement or amputation, may be required for necrotic tissue in severe disease.
All patients with meningococcemia require hospital admission, with intensive care indicated for those with sepsis or shock. Close contacts must receive prompt postexposure prophylaxis, ideally within 24 hours, using agents such as rifampin, ciprofloxacin, or ceftriaxone. Vaccination is recommended for specific high-risk groups and as part of routine adolescent immunization programs.
Key clinical pearls include maintaining a high index of suspicion, notifying public health authorities immediately, and never delaying antibiotic therapy for diagnostic testing. Rapid progression, early shock, and rash should always prompt consideration of meningococcemia, as early treatment is critical to survival.
Meningococcemia is a serious bacterial illness caused by Neisseria meningitidis. It can present in several clinical forms, ranging from mild, self-limited disease to rapidly fatal overwhelming sepsis. Other manifestations include meningococcal meningitis, chronic or occult meningococcemia, septic arthritis, and less commonly localized infections. Transmission occurs through close contact with an infected person or an asymptomatic carrier, with intimate kissing and cigarette smoking recognized as independent risk factors.
The disease is caused by Neisseria meningitidis, a gram-negative diplococcus with multiple serotypes. Although many serotypes exist, most infections are caused by serogroups A, B, C, X, Y, and W135, with serogroup B being the most common in the United States. The organism colonizes the human nasopharynx, the only natural reservoir, and may invade the bloodstream after penetrating nasopharyngeal epithelial cells. Circulating bacteria are normally cleared by the spleen, but meningococci produce a potent endotoxin, lipooligosaccharide, which plays a central role in vascular injury, skin manifestations, adrenal hemorrhage, and circulatory collapse. Carrier rates increase with age, reaching up to 30–40% in young adults, while young children have lower immunity and higher infection risk. Disease incidence peaks in fall and spring and is higher in crowded living conditions such as military barracks and dormitories.
Clinical presentation varies widely. Mild meningococcemia is the most common form and is often preceded by an upper respiratory infection. Patients develop fever, chills, myalgias, arthralgias, and malaise, and the illness may resolve spontaneously over several days. However, this form can progress to meningitis or overwhelming sepsis. Overwhelming meningococcal sepsis occurs in approximately 10% of cases and carries a high mortality rate, with most deaths occurring within the first 48 hours. The onset is abrupt, and early symptoms may appear deceptively mild, including low-grade tachycardia, tachypnea, hypotension, fever, vomiting, headache, rash, and muscle tenderness. Infants may present with lethargy, poor feeding, or a bulging fontanel.
A characteristic rash is a key feature, often beginning as petechiae on the skin, mucous membranes, or conjunctivae, and progressing to purpura and ecchymoses. In severe cases, the rash may coalesce and become necrotic, leading to purpura fulminans. Rapid deterioration may follow, with hypotension, shock, metabolic acidosis, acute respiratory distress syndrome, and disseminated intravascular coagulation. Meningitis may or may not be present. A particularly severe complication is Waterhouse–Friderichsen syndrome, marked by bilateral adrenal hemorrhage, vasomotor collapse, and acute renal failure due to prolonged hypotension.
Chronic meningococcemia is uncommon and presents as a more indolent illness over weeks to months. Patients are often well appearing but experience recurrent fevers, chills, arthralgias, migratory polyarthritis, intermittent painful rashes on the extremities, and sometimes splenomegaly. Meningococcal meningitis presents with headache, fever, neck stiffness, altered mental status, lethargy, or obtundation. Septic arthritis may occur during active bacteremia and typically involves multiple joints with severe pain, swelling, effusion, and restricted movement. Less common manifestations include conjunctivitis, sinusitis, pneumonia, urethritis, salpingitis, prostatitis, myocarditis, and pericarditis.
Diagnosis is primarily clinical and should be suspected in any patient with fever, rash, and rapid clinical deterioration. Importantly, diagnostic evaluation must never delay resuscitation or antibiotic administration. Gram stain and cultures may be obtained from blood, cerebrospinal fluid, joint aspirates, sputum, urine, or scrapings of petechial or papular lesions, which may reveal intracellular or extracellular gram-negative diplococci. Laboratory findings may include leukocytosis early in disease, followed by leukopenia in severe cases, thrombocytopenia in association with purpura or DIC, metabolic acidosis, renal dysfunction, and abnormal coagulation parameters. Polymerase chain reaction testing is particularly useful when antibiotics have already been administered.
Management requires immediate stabilization with strict attention to airway, breathing, and circulation. Droplet precautions should be instituted, and public health authorities notified. Aggressive fluid resuscitation is essential for hypotension, with vasopressors such as dopamine, norepinephrine, or epinephrine if shock persists. Early endotracheal intubation is indicated for severe hypoxia, acidosis, or altered mental status. Empiric intravenous antibiotics must be started promptly, typically with a third-generation cephalosporin, and later tailored once the diagnosis is confirmed. Penicillin remains an option for proven susceptible infections, while chloramphenicol may be used in penicillin-allergic patients.
Supportive care includes management of metabolic acidosis, monitoring of urine output, treatment of DIC with blood products as indicated, and consideration of corticosteroids in select cases, particularly with adrenal involvement. Surgical intervention, including debridement or amputation, may be required for necrotic tissue in severe disease.
All patients with meningococcemia require hospital admission, with intensive care indicated for those with sepsis or shock. Close contacts must receive prompt postexposure prophylaxis, ideally within 24 hours, using agents such as rifampin, ciprofloxacin, or ceftriaxone. Vaccination is recommended for specific high-risk groups and as part of routine adolescent immunization programs.
Key clinical pearls include maintaining a high index of suspicion, notifying public health authorities immediately, and never delaying antibiotic therapy for diagnostic testing. Rapid progression, early shock, and rash should always prompt consideration of meningococcemia, as early treatment is critical to survival.
- Published on
KembaraXtra-Medicine – Methanol Poisoning
Methanol is a colorless, volatile liquid that is rapidly absorbed within 30–60 minutes and metabolized by the liver, with a half-life of approximately 4–8 hours. Methanol itself is relatively nontoxic and produces inebriation similar to ethanol. Toxicity results from its metabolites, formaldehyde and formic acid, which inhibit cytochrome oxidase and disrupt cellular respiration. Formic acid is primarily responsible for severe metabolic acidosis, visual toxicity, and mortality, as it is directly toxic to the retina and optic nerve. Methanol is metabolized in three steps: conversion to formaldehyde by alcohol dehydrogenase, rapid conversion of formaldehyde to formic acid by aldehyde dehydrogenase, and folate-dependent degradation of formic acid to carbon dioxide and water. The first and third steps are rate-limiting.
Common sources of methanol exposure include windshield washer fluid, wood alcohol, carburetor cleaners, fuel antifreeze solutions, formalin, gasoline, paint solvents, household cleaners, Sterno cans, moonshine, model airplane fuel, photocopying fluids, and perfumes. Ingestion may be intentional or accidental, and inhalational exposure can occur with solvent abuse.
Clinical manifestations often begin with gastrointestinal symptoms such as anorexia, nausea, vomiting, and abdominal pain. Central nervous system findings include headache, dizziness, confusion, inebriation, seizures, and coma. Ophthalmologic symptoms are characteristic and include blurry or hazy vision, photophobia, “snowfield” vision, central scotomas, and potentially permanent blindness. Patients may present without a clear ingestion history but instead with an unexplained high anion gap metabolic acidosis and elevated osmol gap. On examination, findings may include tachypnea, altered mental status, optic disc hyperemia or pallor, papilledema, and afferent pupillary defects.
Evaluation requires a careful history of all possible ingestions and attention to visual complaints, along with a funduscopic examination. Essential laboratory tests include arterial blood gas analysis, serum electrolytes, glucose, BUN, creatinine, measured serum osmolality, and serum levels of methanol, ethanol, ethylene glycol, and isopropyl alcohol. The anion gap and osmol gap should be calculated. The osmol gap is most useful early in poisoning, as methanol is osmotically active, whereas its toxic metabolites are not. Importantly, a normal osmol gap does not exclude methanol poisoning, especially in late presenters. Serum methanol levels confirm the diagnosis, though levels may be undetectable late in the course when severe acidosis is present.
Management begins with prompt stabilization of airway, breathing, and circulation, along with administration of dextrose, naloxone, and thiamine for altered mental status. Further absorption is limited by supportive care, as gastric lavage and activated charcoal have limited benefit due to rapid methanol absorption. The cornerstone of treatment is inhibition of alcohol dehydrogenase to prevent formation of toxic metabolites. Fomepizole is the preferred antidote and should be initiated promptly when methanol ingestion is suspected, even before serum levels return. Ethanol infusion is an alternative when fomepizole is unavailable but is associated with significant adverse effects and requires close monitoring.
Hemodialysis plays a critical role by enhancing elimination of methanol and its metabolites and correcting severe acidosis. Indications include visual symptoms, severe or refractory metabolic acidosis, renal failure, ingestion of large amounts of methanol, or serum methanol levels greater than 25 mg/dL. Hemodialysis is continued until methanol levels fall below 25 mg/dL and acidosis resolves. Adjunctive therapy with folic or folinic acid is recommended to enhance metabolism of formic acid. Sodium bicarbonate is used to correct severe acidosis, with the goal of maintaining a normal serum pH.
Patients with significant exposure require hospital admission, often to the intensive care unit, and transfer to a facility with antidote availability and hemodialysis capability if needed. Asymptomatic patients with low methanol levels, normal acid–base status, and stable electrolytes may be considered for discharge. Psychiatric evaluation is important in cases of intentional ingestion. Early recognition and treatment are essential, as delays greatly increase the risk of blindness and death.
Methanol is a colorless, volatile liquid that is rapidly absorbed within 30–60 minutes and metabolized by the liver, with a half-life of approximately 4–8 hours. Methanol itself is relatively nontoxic and produces inebriation similar to ethanol. Toxicity results from its metabolites, formaldehyde and formic acid, which inhibit cytochrome oxidase and disrupt cellular respiration. Formic acid is primarily responsible for severe metabolic acidosis, visual toxicity, and mortality, as it is directly toxic to the retina and optic nerve. Methanol is metabolized in three steps: conversion to formaldehyde by alcohol dehydrogenase, rapid conversion of formaldehyde to formic acid by aldehyde dehydrogenase, and folate-dependent degradation of formic acid to carbon dioxide and water. The first and third steps are rate-limiting.
Common sources of methanol exposure include windshield washer fluid, wood alcohol, carburetor cleaners, fuel antifreeze solutions, formalin, gasoline, paint solvents, household cleaners, Sterno cans, moonshine, model airplane fuel, photocopying fluids, and perfumes. Ingestion may be intentional or accidental, and inhalational exposure can occur with solvent abuse.
Clinical manifestations often begin with gastrointestinal symptoms such as anorexia, nausea, vomiting, and abdominal pain. Central nervous system findings include headache, dizziness, confusion, inebriation, seizures, and coma. Ophthalmologic symptoms are characteristic and include blurry or hazy vision, photophobia, “snowfield” vision, central scotomas, and potentially permanent blindness. Patients may present without a clear ingestion history but instead with an unexplained high anion gap metabolic acidosis and elevated osmol gap. On examination, findings may include tachypnea, altered mental status, optic disc hyperemia or pallor, papilledema, and afferent pupillary defects.
Evaluation requires a careful history of all possible ingestions and attention to visual complaints, along with a funduscopic examination. Essential laboratory tests include arterial blood gas analysis, serum electrolytes, glucose, BUN, creatinine, measured serum osmolality, and serum levels of methanol, ethanol, ethylene glycol, and isopropyl alcohol. The anion gap and osmol gap should be calculated. The osmol gap is most useful early in poisoning, as methanol is osmotically active, whereas its toxic metabolites are not. Importantly, a normal osmol gap does not exclude methanol poisoning, especially in late presenters. Serum methanol levels confirm the diagnosis, though levels may be undetectable late in the course when severe acidosis is present.
Management begins with prompt stabilization of airway, breathing, and circulation, along with administration of dextrose, naloxone, and thiamine for altered mental status. Further absorption is limited by supportive care, as gastric lavage and activated charcoal have limited benefit due to rapid methanol absorption. The cornerstone of treatment is inhibition of alcohol dehydrogenase to prevent formation of toxic metabolites. Fomepizole is the preferred antidote and should be initiated promptly when methanol ingestion is suspected, even before serum levels return. Ethanol infusion is an alternative when fomepizole is unavailable but is associated with significant adverse effects and requires close monitoring.
Hemodialysis plays a critical role by enhancing elimination of methanol and its metabolites and correcting severe acidosis. Indications include visual symptoms, severe or refractory metabolic acidosis, renal failure, ingestion of large amounts of methanol, or serum methanol levels greater than 25 mg/dL. Hemodialysis is continued until methanol levels fall below 25 mg/dL and acidosis resolves. Adjunctive therapy with folic or folinic acid is recommended to enhance metabolism of formic acid. Sodium bicarbonate is used to correct severe acidosis, with the goal of maintaining a normal serum pH.
Patients with significant exposure require hospital admission, often to the intensive care unit, and transfer to a facility with antidote availability and hemodialysis capability if needed. Asymptomatic patients with low methanol levels, normal acid–base status, and stable electrolytes may be considered for discharge. Psychiatric evaluation is important in cases of intentional ingestion. Early recognition and treatment are essential, as delays greatly increase the risk of blindness and death.
- Published on
KembaraXtra – Medicine – Ménière Disease
Ménière disease is a chronic disorder of the inner ear characterized by recurrent, spontaneous episodes of vertigo accompanied by sensorineural hearing loss, tinnitus, and a sensation of aural fullness. The condition most commonly affects one ear, although bilateral involvement may occur in up to 40% of patients. The estimated incidence is approximately 15 per 100,000 people in the United States, with a slight female predominance. Although Ménière disease can occur at any age, it most frequently presents between 40 and 60 years of age. While benign and nonfatal, it can lead to significant morbidity due to the unpredictability and severity of symptoms.
The etiology of Ménière disease is idiopathic, with endolymphatic hydrops considered the primary underlying mechanism. This condition results from impaired drainage of endolymph from the endolymphatic sac and duct, leading to increased pressure within the endolymphatic system. Elevated pressure may cause rupture of the membrane separating potassium-rich endolymph from potassium-poor perilymph, resulting in abnormal stimulation and transient dysfunction of vestibular and cochlear nerve receptors. Additional proposed contributors include structural abnormalities of the endolymphatic system, autoimmune processes, genetic predisposition, remote viral injury, and ischemia of the inner ear. It is important to distinguish Ménière disease from secondary Ménière syndrome caused by conditions such as thyroid disease, syphilis, autoimmune inner ear disease, or medication effects.
Diagnosis is based primarily on clinical features supported by neurotologic evaluation. Definitive diagnosis requires histopathologic confirmation and therefore can only be made postmortem, though modern MRI techniques may provide supportive evidence. Diagnostic criteria include at least two spontaneous episodes of vertigo lasting 20 minutes or longer, at least one documented episode of sensorineural hearing loss on audiometric testing, and the presence of tinnitus or aural fullness in the affected ear. Based on the completeness of findings, cases are categorized as definite, probable, or possible Ménière disease.
Patients typically present with the classic tetrad of vertigo, hearing loss, tinnitus, and aural fullness. Vertigo attacks last from minutes to hours and are frequently associated with nausea and vomiting. Hearing loss is sensorineural, fluctuating, and progressive, often initially affecting low frequencies before potentially involving all frequencies over time. Tinnitus is commonly low-pitched and described as roaring, while aural fullness is perceived as pressure or congestion in the affected ear. Attacks usually reach peak intensity rapidly and resolve gradually, leaving patients fatigued and unsteady for hours to days. Some individuals may be asymptomatic between episodes, while others experience persistent imbalance. Sudden drop attacks without loss of consciousness may also occur.
Physical examination findings vary depending on whether the patient is evaluated during or between attacks. During acute episodes, patients may appear pale, diaphoretic, and distressed. Horizontal nystagmus and impaired hearing are commonly observed. Tuning fork testing often reveals lateralization away from the affected ear on Weber testing, with preserved air conduction on Rinne testing. Romberg testing may demonstrate postural instability, particularly with eyes closed. A thorough neurologic and otologic examination is essential to exclude central nervous system pathology or alternative peripheral ear disorders.
The essential evaluation includes a detailed history and complete neurologic examination. Neuroimaging is warranted when central causes of vertigo are suspected or when focal neurologic deficits are present, particularly in patients with new unilateral hearing loss. Audiometric testing is critical for documenting sensorineural hearing loss, and additional vestibular studies such as caloric testing, electronystagmography, or electrocochleography are typically performed in the outpatient setting. Laboratory investigations are reserved for suspected systemic or secondary causes.
Management in the acute setting focuses on symptom control and exclusion of life-threatening conditions such as stroke. Patients should be protected from falls and maintained in a comfortable position. Intravenous fluids are indicated for dehydration due to vomiting. Benzodiazepines and antiemetics are first-line therapies for acute vertigo and nausea, with antihistamines used as adjuncts. Long-term management aims to reduce the frequency and severity of attacks through dietary sodium restriction, avoidance of triggers such as caffeine and alcohol, diuretics, corticosteroids, and selected transtympanic therapies. Surgical interventions are reserved for patients with severe, refractory disease.
Disposition depends on symptom severity and response to treatment. Admission is indicated for patients with intractable vertigo, persistent vomiting, dehydration, or inability to ambulate safely. Most patients can be discharged once symptoms are controlled, oral intake is tolerated, gait is stable, and neurologic examination is normal. Discharged patients should receive fall precautions and counseling to avoid driving or hazardous activities until symptoms have resolved and sedating medications have been discontinued. Outpatient follow-up with otolaryngology, otology, or neurology is recommended for further evaluation, audiometry, and long-term management.

- Published on
KembaraXtra-Medicine – Mercury Poisoning
Mercury poisoning results from exposure to mercury in one of three primary forms: elemental, inorganic salts, or organic mercury. Mercury exerts its toxic effects by reacting with sulfhydryl groups, leading to enzyme inhibition and disruption of cellular membranes. It also binds to phosphoryl, carboxyl, amide, and amine groups of enzymes, further impairing normal cellular function.
Exposure most commonly occurs through inhalation or ingestion, and less frequently through dermal contact. Occupational and environmental exposures are well recognized and include industries such as chlorine and caustic soda manufacturing, dentistry, photography, taxidermy, mercury mining, and the production of thermometers, batteries, lamps, plastics, paints, pigments, fireworks, and explosives. Contaminated seafood is a major source of organic mercury exposure in the general population.
The clinical presentation varies depending on the form of mercury involved. Elemental mercury exposure most often occurs through inhalation, with symptoms developing within hours. Patients may experience cough and dyspnea that can progress to noncardiogenic pulmonary edema, along with metallic taste, excessive salivation, weakness, nausea, diarrhea, fever, headaches, and visual disturbances. Subcutaneous or intravenous exposure may lead to granulomas, abscesses, or pulmonary embolization. Elemental mercury is relatively nontoxic when ingested orally due to poor gastrointestinal absorption.
Inorganic mercurial salt ingestion causes severe caustic gastrointestinal injury. Symptoms include abdominal pain, nausea, vomiting, diarrhea, sore throat, and a metallic taste. Hemorrhagic gastroenteritis with hematemesis and hematochezia may occur, along with acute tubular necrosis and renal failure. Acrodynia, also known as pink disease, is an idiosyncratic reaction seen mainly in children and is characterized by painful extremities, pink discoloration, and desquamation.
Organic mercury exposure, commonly through contaminated seafood, primarily affects the central nervous system. While mild gastrointestinal symptoms may occur acutely, delayed neurologic toxicity predominates and may take weeks to months to appear. Manifestations include paresthesias, ataxia, paralysis, visual field constriction, dysarthria, hearing loss, progressive cognitive decline, and death. Fetal exposure in utero is associated with particularly severe neurologic outcomes.
Diagnosis relies heavily on a detailed exposure history, including occupational risks and recent seafood consumption. Physical examination focuses on identifying respiratory distress, signs of caustic gastrointestinal injury, and neuropsychiatric abnormalities. Laboratory evaluation may reveal renal dysfunction, but mercury levels in blood or urine can be misleading, especially after recent seafood ingestion. Imaging studies such as chest radiographs may show pulmonary edema or metallic deposits, abdominal radiographs can identify ingested mercury, and head CT may demonstrate cerebellar atrophy in chronic cases.
Management begins with immediate removal from the source of exposure and stabilization of airway, breathing, and circulation. Supportive care includes oxygen, intravenous fluids for hypotension, and treatment of altered mental status with dextrose, thiamine, and naloxone when indicated. Elemental mercury inhalation requires close observation for delayed pulmonary edema. Inorganic mercury ingestion warrants aggressive fluid resuscitation and early treatment of shock or renal failure.
Chelation therapy is indicated for symptomatic patients. Dimercaptosuccinic acid (DMSA) is commonly used for organic and some elemental mercury exposures, while dimercaprol (British anti-Lewisite) is preferred for severe inorganic mercury poisoning. Activated charcoal may be used for recent ingestions. Surgical incision and drainage may be required for subcutaneous mercury deposits.
Hospital admission is recommended for patients with respiratory compromise, renal impairment, caustic gastrointestinal injury, or those requiring chelation therapy. Asymptomatic patients exposed to elemental mercury may be discharged after an adequate observation period. Follow-up includes repeat mercury testing, avoidance of seafood before testing, and referral to medical toxicology. Early recognition, thorough exposure history, and appropriate chelation are critical to reducing morbidity and preventing long-term neurologic sequelae.
Mercury poisoning results from exposure to mercury in one of three primary forms: elemental, inorganic salts, or organic mercury. Mercury exerts its toxic effects by reacting with sulfhydryl groups, leading to enzyme inhibition and disruption of cellular membranes. It also binds to phosphoryl, carboxyl, amide, and amine groups of enzymes, further impairing normal cellular function.
Exposure most commonly occurs through inhalation or ingestion, and less frequently through dermal contact. Occupational and environmental exposures are well recognized and include industries such as chlorine and caustic soda manufacturing, dentistry, photography, taxidermy, mercury mining, and the production of thermometers, batteries, lamps, plastics, paints, pigments, fireworks, and explosives. Contaminated seafood is a major source of organic mercury exposure in the general population.
The clinical presentation varies depending on the form of mercury involved. Elemental mercury exposure most often occurs through inhalation, with symptoms developing within hours. Patients may experience cough and dyspnea that can progress to noncardiogenic pulmonary edema, along with metallic taste, excessive salivation, weakness, nausea, diarrhea, fever, headaches, and visual disturbances. Subcutaneous or intravenous exposure may lead to granulomas, abscesses, or pulmonary embolization. Elemental mercury is relatively nontoxic when ingested orally due to poor gastrointestinal absorption.
Inorganic mercurial salt ingestion causes severe caustic gastrointestinal injury. Symptoms include abdominal pain, nausea, vomiting, diarrhea, sore throat, and a metallic taste. Hemorrhagic gastroenteritis with hematemesis and hematochezia may occur, along with acute tubular necrosis and renal failure. Acrodynia, also known as pink disease, is an idiosyncratic reaction seen mainly in children and is characterized by painful extremities, pink discoloration, and desquamation.
Organic mercury exposure, commonly through contaminated seafood, primarily affects the central nervous system. While mild gastrointestinal symptoms may occur acutely, delayed neurologic toxicity predominates and may take weeks to months to appear. Manifestations include paresthesias, ataxia, paralysis, visual field constriction, dysarthria, hearing loss, progressive cognitive decline, and death. Fetal exposure in utero is associated with particularly severe neurologic outcomes.
Diagnosis relies heavily on a detailed exposure history, including occupational risks and recent seafood consumption. Physical examination focuses on identifying respiratory distress, signs of caustic gastrointestinal injury, and neuropsychiatric abnormalities. Laboratory evaluation may reveal renal dysfunction, but mercury levels in blood or urine can be misleading, especially after recent seafood ingestion. Imaging studies such as chest radiographs may show pulmonary edema or metallic deposits, abdominal radiographs can identify ingested mercury, and head CT may demonstrate cerebellar atrophy in chronic cases.
Management begins with immediate removal from the source of exposure and stabilization of airway, breathing, and circulation. Supportive care includes oxygen, intravenous fluids for hypotension, and treatment of altered mental status with dextrose, thiamine, and naloxone when indicated. Elemental mercury inhalation requires close observation for delayed pulmonary edema. Inorganic mercury ingestion warrants aggressive fluid resuscitation and early treatment of shock or renal failure.
Chelation therapy is indicated for symptomatic patients. Dimercaptosuccinic acid (DMSA) is commonly used for organic and some elemental mercury exposures, while dimercaprol (British anti-Lewisite) is preferred for severe inorganic mercury poisoning. Activated charcoal may be used for recent ingestions. Surgical incision and drainage may be required for subcutaneous mercury deposits.
Hospital admission is recommended for patients with respiratory compromise, renal impairment, caustic gastrointestinal injury, or those requiring chelation therapy. Asymptomatic patients exposed to elemental mercury may be discharged after an adequate observation period. Follow-up includes repeat mercury testing, avoidance of seafood before testing, and referral to medical toxicology. Early recognition, thorough exposure history, and appropriate chelation are critical to reducing morbidity and preventing long-term neurologic sequelae.
- Published on
KembaraXtra-Medicine – Mesenteric Ischemia
Mesenteric ischemia is a serious condition caused by decreased or completely occluded blood flow through the mesenteric vessels, leading to ischemia or infarction of the bowel. It may result from arterial obstruction, venous thrombosis, or low-flow states. Although it accounts for only about 1 in 1,000 hospital admissions and 1–2% of admissions for abdominal pain, it carries a very high mortality rate of 60–70%, especially when diagnosis or treatment is delayed beyond 24 hours. Most cases occur in patients older than 50 years.
Acute mesenteric arterial embolism is the most common cause, accounting for approximately half of acute cases. It typically affects elderly patients and most often arises from cardiac sources such as atrial fibrillation, valvular disease, or ventricular thrombus after myocardial infarction. Emboli usually lodge several centimeters distal to the origin of the superior mesenteric artery, sparing proximal bowel segments. Mesenteric arterial thrombosis accounts for about 15% of cases and usually develops from rupture of an atherosclerotic plaque in patients with chronic mesenteric ischemia, often preceded by longstanding postprandial abdominal pain known as intestinal angina.
Mesenteric venous thrombosis represents 5–15% of cases and tends to have a more subacute presentation. It occurs more often in younger patients with underlying hypercoagulable states, including inherited thrombophilias, malignancy, pregnancy, estrogen therapy, sickle cell disease, sepsis, renal failure on dialysis, or recent trauma. Nonocclusive mesenteric ischemia accounts for 20–30% of cases and is associated with low cardiac output states such as congestive heart failure, sepsis, hypotension, hypovolemia, or recent use of vasopressors. This form is associated with poorer survival. Chronic mesenteric ischemia presents with postprandial abdominal pain, food avoidance, and weight loss. Rare causes include mesenteric arterial dissection, median arcuate ligament syndrome, external compression by tumors, and certain medications such as digitalis, ergotamine, cocaine, and vasopressin.
Clinically, acute mesenteric ischemia classically presents with sudden-onset, severe, diffuse abdominal pain that is out of proportion to physical examination findings. Associated symptoms include nausea, vomiting, diarrhea, and occult gastrointestinal bleeding. Elderly patients may present atypically with altered mental status, tachypnea, or tachycardia. As ischemia progresses to infarction, late findings such as peritoneal signs, abdominal distention, and hypoactive bowel sounds may appear.
Laboratory findings are often nonspecific. Leukocytosis is common, metabolic acidosis may be present, and serum lactate is elevated in most patients with advanced disease, correlating with poor prognosis. Imaging plays a crucial role in diagnosis. Plain abdominal radiographs are often normal early but may show pneumatosis intestinalis or pneumobilia in advanced cases. Multidetector CT angiography has become the imaging modality of choice, allowing visualization of mesenteric vessels and bowel wall changes. Angiography, once the gold standard, is now used selectively for both diagnosis and therapeutic intervention.
Management requires rapid recognition and aggressive treatment. Initial stabilization focuses on airway, breathing, and circulation, with prompt fluid resuscitation. Patients should be kept NPO, a nasogastric tube placed for decompression, and electrolyte abnormalities corrected. Broad-spectrum intravenous antibiotics covering bowel flora are essential. Systemic anticoagulation with heparin is recommended unless contraindicated. Vasoconstrictive medications should be avoided when possible, as they may worsen mesenteric perfusion.
Definitive management depends on the underlying cause and severity. Intra-arterial vasodilators such as papaverine may be administered during angiography for vasospasm or nonocclusive ischemia. Thrombolysis or surgical revascularization may be required for arterial occlusion. Any patient with peritoneal signs requires urgent exploratory laparotomy. All patients with mesenteric ischemia require hospital admission and early surgical consultation, as delays in diagnosis and intervention dramatically increase mortality.
Mesenteric ischemia is a serious condition caused by decreased or completely occluded blood flow through the mesenteric vessels, leading to ischemia or infarction of the bowel. It may result from arterial obstruction, venous thrombosis, or low-flow states. Although it accounts for only about 1 in 1,000 hospital admissions and 1–2% of admissions for abdominal pain, it carries a very high mortality rate of 60–70%, especially when diagnosis or treatment is delayed beyond 24 hours. Most cases occur in patients older than 50 years.
Acute mesenteric arterial embolism is the most common cause, accounting for approximately half of acute cases. It typically affects elderly patients and most often arises from cardiac sources such as atrial fibrillation, valvular disease, or ventricular thrombus after myocardial infarction. Emboli usually lodge several centimeters distal to the origin of the superior mesenteric artery, sparing proximal bowel segments. Mesenteric arterial thrombosis accounts for about 15% of cases and usually develops from rupture of an atherosclerotic plaque in patients with chronic mesenteric ischemia, often preceded by longstanding postprandial abdominal pain known as intestinal angina.
Mesenteric venous thrombosis represents 5–15% of cases and tends to have a more subacute presentation. It occurs more often in younger patients with underlying hypercoagulable states, including inherited thrombophilias, malignancy, pregnancy, estrogen therapy, sickle cell disease, sepsis, renal failure on dialysis, or recent trauma. Nonocclusive mesenteric ischemia accounts for 20–30% of cases and is associated with low cardiac output states such as congestive heart failure, sepsis, hypotension, hypovolemia, or recent use of vasopressors. This form is associated with poorer survival. Chronic mesenteric ischemia presents with postprandial abdominal pain, food avoidance, and weight loss. Rare causes include mesenteric arterial dissection, median arcuate ligament syndrome, external compression by tumors, and certain medications such as digitalis, ergotamine, cocaine, and vasopressin.
Clinically, acute mesenteric ischemia classically presents with sudden-onset, severe, diffuse abdominal pain that is out of proportion to physical examination findings. Associated symptoms include nausea, vomiting, diarrhea, and occult gastrointestinal bleeding. Elderly patients may present atypically with altered mental status, tachypnea, or tachycardia. As ischemia progresses to infarction, late findings such as peritoneal signs, abdominal distention, and hypoactive bowel sounds may appear.
Laboratory findings are often nonspecific. Leukocytosis is common, metabolic acidosis may be present, and serum lactate is elevated in most patients with advanced disease, correlating with poor prognosis. Imaging plays a crucial role in diagnosis. Plain abdominal radiographs are often normal early but may show pneumatosis intestinalis or pneumobilia in advanced cases. Multidetector CT angiography has become the imaging modality of choice, allowing visualization of mesenteric vessels and bowel wall changes. Angiography, once the gold standard, is now used selectively for both diagnosis and therapeutic intervention.
Management requires rapid recognition and aggressive treatment. Initial stabilization focuses on airway, breathing, and circulation, with prompt fluid resuscitation. Patients should be kept NPO, a nasogastric tube placed for decompression, and electrolyte abnormalities corrected. Broad-spectrum intravenous antibiotics covering bowel flora are essential. Systemic anticoagulation with heparin is recommended unless contraindicated. Vasoconstrictive medications should be avoided when possible, as they may worsen mesenteric perfusion.
Definitive management depends on the underlying cause and severity. Intra-arterial vasodilators such as papaverine may be administered during angiography for vasospasm or nonocclusive ischemia. Thrombolysis or surgical revascularization may be required for arterial occlusion. Any patient with peritoneal signs requires urgent exploratory laparotomy. All patients with mesenteric ischemia require hospital admission and early surgical consultation, as delays in diagnosis and intervention dramatically increase mortality.
- Published on
KembaraXtra-Medicine – Marine Envenomation
Marine envenomation refers to poisoning caused by the sting or bite of marine vertebrate or invertebrate species. These injuries occur in a wide variety of aquatic environments and range from mild local reactions to rapidly fatal systemic toxicity. Common causative organisms include sponges, jellyfish, echinoderms, mollusks, fish, and sea snakes, each producing distinct clinical syndromes based on venom type and mechanism of delivery.
The etiology varies by species. Sponges contain sharp spicules coated with irritants that penetrate the skin and cause dermatitis. Jellyfish and other cnidarians possess nematocysts that eject hollow toxin-filled tubules on contact; box jellyfish are particularly dangerous and can cause death within minutes. Starfish and sea urchins deliver venom through rigid or hollow spines, while sea cucumbers secrete holothurin, a toxin capable of causing severe ocular injury. Cone shells inject potent neurotoxic peptides through dart-like teeth that interfere with neuromuscular transmission. Stingrays, the most common cause of marine envenomation, inject venom via serrated tail spines. Scorpion fish, including lionfish and stonefish, possess dorsal spines with neurotoxic venom, and catfish envenomate through dorsal and pectoral spines. Sea snakes inject highly neurotoxic venom through hollow fangs, leading to neuromuscular blockade.
Clinical presentation depends on the organism involved and the severity of envenomation. Sponge exposure typically causes delayed itching, burning, local swelling, and systemic symptoms such as fever, malaise, nausea, and muscle cramps, with possible skin desquamation weeks later. Jellyfish stings cause immediate pain, burning, wheals, and blistering, while severe cases may progress to neurologic deficits, cardiovascular collapse, respiratory failure, gastrointestinal symptoms, or ocular injury. Echinoderm injuries range from localized pain and edema with starfish to intense pain, muscle aches, hypotension, and respiratory distress with multiple sea urchin stings. Sea cucumber exposure often causes mild dermatitis but may result in severe corneal injury and blindness.
Mollusk envenomation, particularly from cone shells, begins with sharp puncture pain and paresthesias that may progress to paralysis, respiratory failure, dysphagia, syncope, or disseminated intravascular coagulation. Stingray injuries present as puncture wounds or lacerations with severe pain, edema, bleeding, and possible tissue necrosis, along with systemic symptoms such as nausea, hypotension, dysrhythmias, seizures, and paralysis. Scorpion fish stings cause intense pain lasting hours and may be accompanied by fever, delirium, seizures, and hypertension. Catfish injuries result in local pain, swelling, bleeding, muscle spasms, and neuropathic symptoms. Sea snake bites are often initially painless but progress within minutes to hours to muscle pain, paralysis, trismus, dysphagia, renal failure, and coma, with high mortality if untreated.
Diagnosis relies primarily on a thorough history and physical examination. Important historical elements include the time and location of exposure, type of water, activity at the time of injury, geographic location, symptom onset, and mental status changes. Physical examination focuses on airway stability, vital signs, neurologic status, cardiopulmonary findings, and careful inspection of wounds for retained foreign bodies, cellulitis, or blistering. Laboratory evaluation may include complete blood count, electrolytes, renal and liver function tests, urinalysis, and arterial blood gases in severe cases. Soft tissue radiographs may be useful to identify retained spines or foreign bodies.
Initial management begins in the prehospital setting with removal from the water, airway and breathing support, hemorrhage control, and avoidance of further venom activation. In the emergency setting, stabilization of airway, breathing, and circulation is the priority, with intravenous access and fluid resuscitation as needed. Clinicians must be prepared to manage anaphylaxis, respiratory failure, and severe pain. General measures include tetanus prophylaxis, antihistamines for pruritus, corticosteroids for severe reactions, narcotic analgesia, and antibiotics for deep, contaminated, or high-risk wounds.
Definitive treatment is species-specific. Spicule injuries from sponges are managed by drying the skin, removing spicules with adhesive tape, and soaking with vinegar or isopropyl alcohol. Jellyfish stings should be rinsed with seawater, treated with vinegar to inactivate nematocysts, and carefully debrided; box jellyfish stings require urgent antivenom. Echinoderm, cone shell, stingray, scorpion fish, and catfish injuries are treated with nonscalding hot water immersion for pain relief and venom inactivation, along with wound exploration and spine removal. Stonefish and sea snake envenomations require specific antivenoms, and sea snake bites necessitate early immobilization, pressure bandaging, and preparation for assisted ventilation.
Patients with systemic toxicity, severe local injury, or need for antivenom require hospital admission. Those without systemic symptoms after adequate observation may be discharged with wound care instructions and follow-up. Key clinical pearls include recognizing that most marine toxins are inactivated by heat or acidic solutions, avoiding fresh water on jellyfish stings, and securing antivenom early when indicated, as supplies are limited and delays can be fatal.
Marine envenomation refers to poisoning caused by the sting or bite of marine vertebrate or invertebrate species. These injuries occur in a wide variety of aquatic environments and range from mild local reactions to rapidly fatal systemic toxicity. Common causative organisms include sponges, jellyfish, echinoderms, mollusks, fish, and sea snakes, each producing distinct clinical syndromes based on venom type and mechanism of delivery.
The etiology varies by species. Sponges contain sharp spicules coated with irritants that penetrate the skin and cause dermatitis. Jellyfish and other cnidarians possess nematocysts that eject hollow toxin-filled tubules on contact; box jellyfish are particularly dangerous and can cause death within minutes. Starfish and sea urchins deliver venom through rigid or hollow spines, while sea cucumbers secrete holothurin, a toxin capable of causing severe ocular injury. Cone shells inject potent neurotoxic peptides through dart-like teeth that interfere with neuromuscular transmission. Stingrays, the most common cause of marine envenomation, inject venom via serrated tail spines. Scorpion fish, including lionfish and stonefish, possess dorsal spines with neurotoxic venom, and catfish envenomate through dorsal and pectoral spines. Sea snakes inject highly neurotoxic venom through hollow fangs, leading to neuromuscular blockade.
Clinical presentation depends on the organism involved and the severity of envenomation. Sponge exposure typically causes delayed itching, burning, local swelling, and systemic symptoms such as fever, malaise, nausea, and muscle cramps, with possible skin desquamation weeks later. Jellyfish stings cause immediate pain, burning, wheals, and blistering, while severe cases may progress to neurologic deficits, cardiovascular collapse, respiratory failure, gastrointestinal symptoms, or ocular injury. Echinoderm injuries range from localized pain and edema with starfish to intense pain, muscle aches, hypotension, and respiratory distress with multiple sea urchin stings. Sea cucumber exposure often causes mild dermatitis but may result in severe corneal injury and blindness.
Mollusk envenomation, particularly from cone shells, begins with sharp puncture pain and paresthesias that may progress to paralysis, respiratory failure, dysphagia, syncope, or disseminated intravascular coagulation. Stingray injuries present as puncture wounds or lacerations with severe pain, edema, bleeding, and possible tissue necrosis, along with systemic symptoms such as nausea, hypotension, dysrhythmias, seizures, and paralysis. Scorpion fish stings cause intense pain lasting hours and may be accompanied by fever, delirium, seizures, and hypertension. Catfish injuries result in local pain, swelling, bleeding, muscle spasms, and neuropathic symptoms. Sea snake bites are often initially painless but progress within minutes to hours to muscle pain, paralysis, trismus, dysphagia, renal failure, and coma, with high mortality if untreated.
Diagnosis relies primarily on a thorough history and physical examination. Important historical elements include the time and location of exposure, type of water, activity at the time of injury, geographic location, symptom onset, and mental status changes. Physical examination focuses on airway stability, vital signs, neurologic status, cardiopulmonary findings, and careful inspection of wounds for retained foreign bodies, cellulitis, or blistering. Laboratory evaluation may include complete blood count, electrolytes, renal and liver function tests, urinalysis, and arterial blood gases in severe cases. Soft tissue radiographs may be useful to identify retained spines or foreign bodies.
Initial management begins in the prehospital setting with removal from the water, airway and breathing support, hemorrhage control, and avoidance of further venom activation. In the emergency setting, stabilization of airway, breathing, and circulation is the priority, with intravenous access and fluid resuscitation as needed. Clinicians must be prepared to manage anaphylaxis, respiratory failure, and severe pain. General measures include tetanus prophylaxis, antihistamines for pruritus, corticosteroids for severe reactions, narcotic analgesia, and antibiotics for deep, contaminated, or high-risk wounds.
Definitive treatment is species-specific. Spicule injuries from sponges are managed by drying the skin, removing spicules with adhesive tape, and soaking with vinegar or isopropyl alcohol. Jellyfish stings should be rinsed with seawater, treated with vinegar to inactivate nematocysts, and carefully debrided; box jellyfish stings require urgent antivenom. Echinoderm, cone shell, stingray, scorpion fish, and catfish injuries are treated with nonscalding hot water immersion for pain relief and venom inactivation, along with wound exploration and spine removal. Stonefish and sea snake envenomations require specific antivenoms, and sea snake bites necessitate early immobilization, pressure bandaging, and preparation for assisted ventilation.
Patients with systemic toxicity, severe local injury, or need for antivenom require hospital admission. Those without systemic symptoms after adequate observation may be discharged with wound care instructions and follow-up. Key clinical pearls include recognizing that most marine toxins are inactivated by heat or acidic solutions, avoiding fresh water on jellyfish stings, and securing antivenom early when indicated, as supplies are limited and delays can be fatal.

- Published on
KembaraXtra-Medicine – Methanol Poisoning
Methanol is a colorless, volatile liquid that is rapidly absorbed within 30–60 minutes and metabolized by the liver, with a half-life of approximately 4–8 hours. Methanol itself is relatively nontoxic and produces inebriation similar to ethanol. Toxicity results from its metabolites, formaldehyde and formic acid, which inhibit cytochrome oxidase and disrupt cellular respiration. Formic acid is primarily responsible for severe metabolic acidosis, visual toxicity, and mortality, as it is directly toxic to the retina and optic nerve. Methanol is metabolized in three steps: conversion to formaldehyde by alcohol dehydrogenase, rapid conversion of formaldehyde to formic acid by aldehyde dehydrogenase, and folate-dependent degradation of formic acid to carbon dioxide and water. The first and third steps are rate-limiting.
Common sources of methanol exposure include windshield washer fluid, wood alcohol, carburetor cleaners, fuel antifreeze solutions, formalin, gasoline, paint solvents, household cleaners, Sterno cans, moonshine, model airplane fuel, photocopying fluids, and perfumes. Ingestion may be intentional or accidental, and inhalational exposure can occur with solvent abuse.
Clinical manifestations often begin with gastrointestinal symptoms such as anorexia, nausea, vomiting, and abdominal pain. Central nervous system findings include headache, dizziness, confusion, inebriation, seizures, and coma. Ophthalmologic symptoms are characteristic and include blurry or hazy vision, photophobia, “snowfield” vision, central scotomas, and potentially permanent blindness. Patients may present without a clear ingestion history but instead with an unexplained high anion gap metabolic acidosis and elevated osmol gap. On examination, findings may include tachypnea, altered mental status, optic disc hyperemia or pallor, papilledema, and afferent pupillary defects.
Evaluation requires a careful history of all possible ingestions and attention to visual complaints, along with a funduscopic examination. Essential laboratory tests include arterial blood gas analysis, serum electrolytes, glucose, BUN, creatinine, measured serum osmolality, and serum levels of methanol, ethanol, ethylene glycol, and isopropyl alcohol. The anion gap and osmol gap should be calculated. The osmol gap is most useful early in poisoning, as methanol is osmotically active, whereas its toxic metabolites are not. Importantly, a normal osmol gap does not exclude methanol poisoning, especially in late presenters. Serum methanol levels confirm the diagnosis, though levels may be undetectable late in the course when severe acidosis is present.
Management begins with prompt stabilization of airway, breathing, and circulation, along with administration of dextrose, naloxone, and thiamine for altered mental status. Further absorption is limited by supportive care, as gastric lavage and activated charcoal have limited benefit due to rapid methanol absorption. The cornerstone of treatment is inhibition of alcohol dehydrogenase to prevent formation of toxic metabolites. Fomepizole is the preferred antidote and should be initiated promptly when methanol ingestion is suspected, even before serum levels return. Ethanol infusion is an alternative when fomepizole is unavailable but is associated with significant adverse effects and requires close monitoring.
Hemodialysis plays a critical role by enhancing elimination of methanol and its metabolites and correcting severe acidosis. Indications include visual symptoms, severe or refractory metabolic acidosis, renal failure, ingestion of large amounts of methanol, or serum methanol levels greater than 25 mg/dL. Hemodialysis is continued until methanol levels fall below 25 mg/dL and acidosis resolves. Adjunctive therapy with folic or folinic acid is recommended to enhance metabolism of formic acid. Sodium bicarbonate is used to correct severe acidosis, with the goal of maintaining a normal serum pH.
Patients with significant exposure require hospital admission, often to the intensive care unit, and transfer to a facility with antidote availability and hemodialysis capability if needed. Asymptomatic patients with low methanol levels, normal acid–base status, and stable electrolytes may be considered for discharge. Psychiatric evaluation is important in cases of intentional ingestion. Early recognition and treatment are essential, as delays greatly increase the risk of blindness and death.
Methanol is a colorless, volatile liquid that is rapidly absorbed within 30–60 minutes and metabolized by the liver, with a half-life of approximately 4–8 hours. Methanol itself is relatively nontoxic and produces inebriation similar to ethanol. Toxicity results from its metabolites, formaldehyde and formic acid, which inhibit cytochrome oxidase and disrupt cellular respiration. Formic acid is primarily responsible for severe metabolic acidosis, visual toxicity, and mortality, as it is directly toxic to the retina and optic nerve. Methanol is metabolized in three steps: conversion to formaldehyde by alcohol dehydrogenase, rapid conversion of formaldehyde to formic acid by aldehyde dehydrogenase, and folate-dependent degradation of formic acid to carbon dioxide and water. The first and third steps are rate-limiting.
Common sources of methanol exposure include windshield washer fluid, wood alcohol, carburetor cleaners, fuel antifreeze solutions, formalin, gasoline, paint solvents, household cleaners, Sterno cans, moonshine, model airplane fuel, photocopying fluids, and perfumes. Ingestion may be intentional or accidental, and inhalational exposure can occur with solvent abuse.
Clinical manifestations often begin with gastrointestinal symptoms such as anorexia, nausea, vomiting, and abdominal pain. Central nervous system findings include headache, dizziness, confusion, inebriation, seizures, and coma. Ophthalmologic symptoms are characteristic and include blurry or hazy vision, photophobia, “snowfield” vision, central scotomas, and potentially permanent blindness. Patients may present without a clear ingestion history but instead with an unexplained high anion gap metabolic acidosis and elevated osmol gap. On examination, findings may include tachypnea, altered mental status, optic disc hyperemia or pallor, papilledema, and afferent pupillary defects.
Evaluation requires a careful history of all possible ingestions and attention to visual complaints, along with a funduscopic examination. Essential laboratory tests include arterial blood gas analysis, serum electrolytes, glucose, BUN, creatinine, measured serum osmolality, and serum levels of methanol, ethanol, ethylene glycol, and isopropyl alcohol. The anion gap and osmol gap should be calculated. The osmol gap is most useful early in poisoning, as methanol is osmotically active, whereas its toxic metabolites are not. Importantly, a normal osmol gap does not exclude methanol poisoning, especially in late presenters. Serum methanol levels confirm the diagnosis, though levels may be undetectable late in the course when severe acidosis is present.
Management begins with prompt stabilization of airway, breathing, and circulation, along with administration of dextrose, naloxone, and thiamine for altered mental status. Further absorption is limited by supportive care, as gastric lavage and activated charcoal have limited benefit due to rapid methanol absorption. The cornerstone of treatment is inhibition of alcohol dehydrogenase to prevent formation of toxic metabolites. Fomepizole is the preferred antidote and should be initiated promptly when methanol ingestion is suspected, even before serum levels return. Ethanol infusion is an alternative when fomepizole is unavailable but is associated with significant adverse effects and requires close monitoring.
Hemodialysis plays a critical role by enhancing elimination of methanol and its metabolites and correcting severe acidosis. Indications include visual symptoms, severe or refractory metabolic acidosis, renal failure, ingestion of large amounts of methanol, or serum methanol levels greater than 25 mg/dL. Hemodialysis is continued until methanol levels fall below 25 mg/dL and acidosis resolves. Adjunctive therapy with folic or folinic acid is recommended to enhance metabolism of formic acid. Sodium bicarbonate is used to correct severe acidosis, with the goal of maintaining a normal serum pH.
Patients with significant exposure require hospital admission, often to the intensive care unit, and transfer to a facility with antidote availability and hemodialysis capability if needed. Asymptomatic patients with low methanol levels, normal acid–base status, and stable electrolytes may be considered for discharge. Psychiatric evaluation is important in cases of intentional ingestion. Early recognition and treatment are essential, as delays greatly increase the risk of blindness and death.

