- Published on
KembaraXtra – Medicine – Ménière Disease
Ménière disease is a chronic disorder of the inner ear characterized by recurrent, spontaneous episodes of vertigo accompanied by sensorineural hearing loss, tinnitus, and a sensation of aural fullness. The condition most commonly affects one ear, although bilateral involvement may occur in up to 40% of patients. The estimated incidence is approximately 15 per 100,000 people in the United States, with a slight female predominance. Although Ménière disease can occur at any age, it most frequently presents between 40 and 60 years of age. While benign and nonfatal, it can lead to significant morbidity due to the unpredictability and severity of symptoms.
The etiology of Ménière disease is idiopathic, with endolymphatic hydrops considered the primary underlying mechanism. This condition results from impaired drainage of endolymph from the endolymphatic sac and duct, leading to increased pressure within the endolymphatic system. Elevated pressure may cause rupture of the membrane separating potassium-rich endolymph from potassium-poor perilymph, resulting in abnormal stimulation and transient dysfunction of vestibular and cochlear nerve receptors. Additional proposed contributors include structural abnormalities of the endolymphatic system, autoimmune processes, genetic predisposition, remote viral injury, and ischemia of the inner ear. It is important to distinguish Ménière disease from secondary Ménière syndrome caused by conditions such as thyroid disease, syphilis, autoimmune inner ear disease, or medication effects.
Diagnosis is based primarily on clinical features supported by neurotologic evaluation. Definitive diagnosis requires histopathologic confirmation and therefore can only be made postmortem, though modern MRI techniques may provide supportive evidence. Diagnostic criteria include at least two spontaneous episodes of vertigo lasting 20 minutes or longer, at least one documented episode of sensorineural hearing loss on audiometric testing, and the presence of tinnitus or aural fullness in the affected ear. Based on the completeness of findings, cases are categorized as definite, probable, or possible Ménière disease.
Patients typically present with the classic tetrad of vertigo, hearing loss, tinnitus, and aural fullness. Vertigo attacks last from minutes to hours and are frequently associated with nausea and vomiting. Hearing loss is sensorineural, fluctuating, and progressive, often initially affecting low frequencies before potentially involving all frequencies over time. Tinnitus is commonly low-pitched and described as roaring, while aural fullness is perceived as pressure or congestion in the affected ear. Attacks usually reach peak intensity rapidly and resolve gradually, leaving patients fatigued and unsteady for hours to days. Some individuals may be asymptomatic between episodes, while others experience persistent imbalance. Sudden drop attacks without loss of consciousness may also occur.
Physical examination findings vary depending on whether the patient is evaluated during or between attacks. During acute episodes, patients may appear pale, diaphoretic, and distressed. Horizontal nystagmus and impaired hearing are commonly observed. Tuning fork testing often reveals lateralization away from the affected ear on Weber testing, with preserved air conduction on Rinne testing. Romberg testing may demonstrate postural instability, particularly with eyes closed. A thorough neurologic and otologic examination is essential to exclude central nervous system pathology or alternative peripheral ear disorders.
The essential evaluation includes a detailed history and complete neurologic examination. Neuroimaging is warranted when central causes of vertigo are suspected or when focal neurologic deficits are present, particularly in patients with new unilateral hearing loss. Audiometric testing is critical for documenting sensorineural hearing loss, and additional vestibular studies such as caloric testing, electronystagmography, or electrocochleography are typically performed in the outpatient setting. Laboratory investigations are reserved for suspected systemic or secondary causes.
Management in the acute setting focuses on symptom control and exclusion of life-threatening conditions such as stroke. Patients should be protected from falls and maintained in a comfortable position. Intravenous fluids are indicated for dehydration due to vomiting. Benzodiazepines and antiemetics are first-line therapies for acute vertigo and nausea, with antihistamines used as adjuncts. Long-term management aims to reduce the frequency and severity of attacks through dietary sodium restriction, avoidance of triggers such as caffeine and alcohol, diuretics, corticosteroids, and selected transtympanic therapies. Surgical interventions are reserved for patients with severe, refractory disease.
Disposition depends on symptom severity and response to treatment. Admission is indicated for patients with intractable vertigo, persistent vomiting, dehydration, or inability to ambulate safely. Most patients can be discharged once symptoms are controlled, oral intake is tolerated, gait is stable, and neurologic examination is normal. Discharged patients should receive fall precautions and counseling to avoid driving or hazardous activities until symptoms have resolved and sedating medications have been discontinued. Outpatient follow-up with otolaryngology, otology, or neurology is recommended for further evaluation, audiometry, and long-term management.

- Published on
Emergency and Acute Medicine – Meckel Diverticulum
Basics description
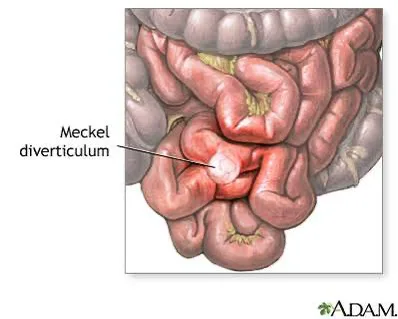
Meckel diverticulum is the most common congenital abnormality of the gastrointestinal tract and results from incomplete obliteration of the omphalomesenteric duct. It is a true diverticulum containing all layers of the bowel wall. Approximately half contain normal ileal mucosa, while the remainder contain ectopic tissue, most commonly gastric, but also pancreatic, duodenal, colonic, endometrial, or hepatobiliary mucosa. The classic “rule of 2’s” applies: it occurs in about 2% of the population, carries a 2% lifetime risk of complications that decreases with age, is usually around 2 inches long, and is located within 2 feet of the ileocecal valve. Symptoms commonly occur around 2 years of age, with nearly half of symptomatic patients presenting before age 2. Although prevalence is similar in males and females, males are more often symptomatic. Complications differ by age, with obstruction and diverticulitis more common in adults and hemorrhage and obstruction more common in children. The mean age of presentation is about 10 years, and current mortality is extremely low.
Etiology
Meckel diverticulum arises from a remnant of the omphalomesenteric duct, which normally regresses by the seventh week of gestation.
Pathophysiology and complications
Obstruction can occur when the diverticulum is attached to the umbilicus, abdominal wall, or other viscera, or when it is free and acts as a lead point. This may result in intussusception, with the diverticulum serving as the leading edge, or volvulus caused by a persistent fibrous band that allows bowel rotation. Diverticulitis occurs when the opening of the diverticulum becomes obstructed, followed by bacterial infection, and often presents similarly to acute appendicitis, which is the most common preoperative diagnosis.
Pediatric considerations
Meckel diverticulum is the most common cause of significant lower gastrointestinal bleeding in children. It typically presents before 5 years of age with episodic, painless, brisk, bright-red rectal bleeding.
Diagnosis, signs, and symptoms
Patients generally present in one of three ways. Rectal bleeding occurs due to hemorrhage from mucosal ulceration associated with ectopic gastric tissue. Vomiting may result from bowel obstruction caused by volvulus, intussusception, or intraperitoneal bands. Abdominal pain, often appendicitis-like, may occur with an inflamed or perforated diverticulum. Associated findings include fever, malaise, weakness, fatigue, abdominal distention, changes in bowel habits, hematochezia or melena, and in advanced cases, peritonitis or septic shock. Tachycardia and hypotension may be present due to pain or blood loss.
Essential workup
Meckel diverticulum can produce a wide variety of nonspecific symptoms, and fewer than 10% of cases are diagnosed preoperatively. It should be considered in patients with recurrent nonspecific abdominal pain, nausea and vomiting, or rectal bleeding. History and physical examination may narrow the diagnosis but are not definitive. Rectal examination is mandatory, and nasogastric tube placement may help rule out an upper gastrointestinal bleed.
Diagnostic tests and interpretation
Laboratory evaluation may reveal a decreased hematocrit from bleeding or leukocytosis in cases of diverticulitis, perforation, or gangrene. Electrolytes, renal function, and coagulation studies should be obtained, and type and screen or cross-match is required with significant bleeding. Computed tomography of the abdomen and pelvis is useful for suspected infection or bowel obstruction but cannot reliably diagnose Meckel diverticulum. Abdominal radiographs may screen for obstruction but are not diagnostic. A technetium-99m pertechnetate scan (Meckel scan) can identify heterotopic gastric mucosa and is highly accurate in children but less sensitive in adults. Small bowel enteroclysis, barium enema, angiography, ultrasound, or laparoscopic evaluation may be used in selected cases. Colonoscopy is not helpful.
Differential diagnosis
In adults, the differential includes appendicitis, adhesions, bowel obstruction, diverticulitis, hemorrhoids, inflammatory bowel disease, internal hernias, intussusception, peptic ulcer disease, pseudomembranous colitis, and volvulus. In children, considerations include anal fissures, appendicitis, atresia, gastroenteritis, hemolytic–uremic syndrome, Henoch–Schönlein purpura, intussusception, malrotation, milk allergy, strictures, and volvulus.
Treatment and emergency management
Prehospital management includes establishing intravenous access for patients with rectal bleeding or abdominal pain. Initial stabilization focuses on resuscitation and early surgical consultation. Hypotension is treated with aggressive fluid resuscitation, packed red blood cell transfusion for brisk bleeding, and vasopressors if septic shock is present. In the emergency department, gastrointestinal bleeding is managed with fluids and transfusion as indicated, Foley catheter placement to monitor urine output, and nasogastric tube placement to exclude brisk upper gastrointestinal bleeding. Obstruction requires nasogastric decompression and surgical consultation. Suspected diverticulitis or perforation requires nil per os status, preoperative antibiotics, and urgent surgical evaluation.
Definitive management
Symptomatic Meckel diverticula should be surgically resected. In children, asymptomatic Meckel diverticula discovered incidentally during laparotomy are generally resected to prevent future complications.
Disposition and follow-up
Admission is required for presumptive or confirmed Meckel diverticulum associated with diverticulitis, obstruction, intussusception, hemorrhage, or volvulus. There are no discharge criteria from the emergency department for suspected cases. Postoperative surgical follow-up is required.
Pearls and pitfalls
Painless, brisk, bright-red rectal bleeding in an infant is most often caused by Meckel diverticulum. The condition presents with a broad range of complications, including obstruction, intussusception, and hemorrhage, and is frequently diagnosed intraoperatively during surgery for presumed appendicitis. Remember the rule of 2’s to aid clinical recognition.
- Published on
Emergency and Acute Medicine – Mastitis
Basics description
Mastitis is an infection of the breast characterized by pain, swelling, erythema, and warmth, most commonly occurring in women who are breast-feeding. Systemic symptoms such as malaise and fever are frequent. Incidence may reach up to one-third of lactating women, with onset typically between 2–3 weeks and several months postpartum, most often before the infant is 3 months old. It is uncommon during the first postpartum week and is more frequent in advanced maternal age and patients with diabetes. Potential complications include recurrence, abscess formation, sepsis, necrotizing fasciitis, fistula, scarring, and breast hypoplasia. In rare cases, mastitis can occur in full-term infants younger than 2 months.
Etiology and risk factors
Staphylococcus aureus is the most common causative organism. Less frequent pathogens include coagulase-negative Staphylococcus, Streptococcus species, Escherichia coli, Haemophilus influenzae, and Candida albicans. Predisposing factors include cracked nipples, poor infant latch, local milk stasis, nipple piercing, poor maternal nutrition, prior mastitis, primiparity, tight bras, sore nipples, short infant frenulum, use of manual breast pumps, and maternal yeast infection.
Diagnosis signs and symptoms
Patients typically present with fever and chills, often exceeding 38.3°C, along with generalized malaise and tachycardia. Local findings include unilateral breast pain, induration, erythema, warmth, and swelling, sometimes in a wedge-shaped distribution. Decreased milk outflow and purulent nipple discharge may occur. Axillary lymphadenopathy can be present.
Essential workup and diagnostic testing
Diagnosis is primarily clinical. Careful breast examination is essential to assess for abscess formation, which may be subtle and is often periareolar. Breast milk cultures are generally unnecessary. Ultrasound should be obtained if abscess is suspected, while mammography is not indicated acutely. In neonates, evaluation for abscess and systemic infection is critical, and a sepsis workup may be required if the infant is febrile or ill appearing.
Differential diagnosis
Conditions to consider include breast engorgement, which is typically bilateral and occurs early postpartum with transient low-grade fever, inflammatory breast carcinoma, cysts or tumors, and breast abscess.
Treatment and emergency management
Breast-feeding should be continued whenever possible, as milk drainage is therapeutic and does not harm the infant. If the infant refuses milk from the affected breast, pumping and discarding milk is recommended. Supportive care includes breast massage, warm or cold compresses, hydration, and optimization of breast-feeding technique, often with lactation consultant support. Mild early cases may resolve without antibiotics. When antibiotics are required, oral therapy for 7–14 days is standard, with dicloxacillin or a first-generation cephalosporin as first-line options. Clindamycin, trimethoprim/sulfamethoxazole, or erythromycin may be used in penicillin-allergic patients or when MRSA is suspected, guided by local resistance patterns. Surgical consultation is indicated for suspected abscess.
Disposition and follow-up
Most patients can be managed as outpatients, with clinical improvement expected within 48 hours of therapy. Admission is indicated for patients requiring incision and drainage under anesthesia, those who are immunocompromised or septic, patients with diabetes and severe infection, and neonates with mastitis. Follow-up with a primary care physician is recommended, and lactation consultant involvement is often beneficial. Persistent or atypical cases should be evaluated to exclude inflammatory breast carcinoma.
Key points and cautions
Discontinuation of breast-feeding increases milk stasis and the risk of abscess formation. Many cases respond to supportive care alone, and unnecessary cessation of breast-feeding is a common and avoidable complication.

- Published on
Emergency and Acute Medicine – Mastoiditis
Basics description
Mastoiditis is inflammation of the mastoid air cells of the temporal bone, most commonly resulting from direct extension of acute purulent otitis media. The middle ear and mastoid air cells communicate through the aditus to the mastoid antrum. In otitis media, inflammatory obstruction of this channel leads to fluid trapping, creating an environment for infection. Clinical manifestations range from mild inflammation of mastoid air cells to aggressive infection with bone destruction and serious intracranial or extracranial complications.
Acute mastoiditis occurs to some degree in all cases of otitis media, with early signs indistinguishable from acute otitis media. As infection progresses, involvement of the periosteum causes periostitis, which may be associated with a subperiosteal abscess. Further progression leads to acute mastoid ostitis (coalescent mastoiditis), characterized by destruction of mastoid trabeculae and coalescence of air cells, sometimes with mastoid empyema or a draining fistula. Untreated disease may result in severe head and neck or intracranial complications.
Masked mastoiditis refers to persistent mastoid infection after partial treatment of otitis media, which may later progress to acute or coalescent mastoiditis. Chronic mastoiditis is defined as infection lasting longer than 3 months. Mastoiditis may also complicate systemic conditions such as leukemia, infectious mononucleosis, sarcoma of the temporal bone, HIV, or Kawasaki disease. Although less common in the antibiotic era, it remains more frequent in infants and young children.
Etiology
Pathogens are similar to those causing otitis media but differ in prevalence. Common organisms in acute mastoiditis include Streptococcus pneumoniae, group A streptococcus, Staphylococcus aureus, and Haemophilus influenzae. Chronic mastoiditis more often involves gram-negative enteric organisms such as Pseudomonas aeruginosa, Escherichia coli, Proteus mirabilis, and Bacteroides species. Less common causes include Mycobacterium tuberculosis and Aspergillus species in immunocompromised patients. In children, S. pneumoniae remains the most frequent pathogen.
Diagnosis signs and symptoms
Patients commonly present with ear pain, otorrhea, fever, headache, and hearing loss of varying severity. Children may show irritability and a history of recurrent otitis media. Physical examination typically reveals postauricular tenderness, edema, and erythema; lateral and inferior displacement of the auricle; loss of the postauricular crease; swelling of the posterior and superior ear canal wall; and tympanic membrane abnormalities consistent with severe otitis media, including bulging or purulent drainage.
Essential workup and diagnostic testing
Mastoiditis is primarily a clinical diagnosis. Laboratory studies may show leukocytosis. Cultures of otorrhea or surgical drainage are important because of pathogen diversity, and blood cultures should be obtained in toxic-appearing patients. Imaging supports diagnosis and evaluates complications: plain mastoid radiographs may show haziness or opacification but have low sensitivity. CT of the temporal bone is more useful, especially for detecting abscesses, trabecular destruction, and complications. MRI is reserved for suspected intracranial involvement not clarified by CT. Lumbar puncture is indicated if meningitis is suspected. In children, judicious use of CT is recommended to minimize radiation exposure when clinical diagnosis is clear.
Differential diagnosis
Otitis media, cellulitis, otitis externa, scalp infection with posterior auricular lymphadenitis, rubella, trauma to the pinna or postauricular area, and meningitis.
Treatment and emergency management
Initial management follows standard ABCs, with IV fluids for hypotension or dehydration. Prompt initiation of IV antibiotics is essential. Given the increasing role of S. aureus, empiric regimens should include antistaphylococcal coverage until culture results are available, with consideration of antipseudomonal coverage in chronic disease. Otolaryngology consultation is mandatory. Surgical drainage is the definitive therapy for acute or coalescent mastoiditis and is emergent in toxic patients. Procedures may include myringotomy with tympanostomy tube placement or mastoidectomy with drainage, required in approximately half of cases. Analgesia with NSAIDs or opioids should be provided.
Disposition and follow-up
All patients with suspected acute or coalescent mastoiditis require admission. Discharge is not appropriate in these cases. Otolaryngology follow-up is essential, and audiography should be performed after resolution to assess for hearing loss.
Complications
Potential complications include Bezold abscess, petrositis, calvarial osteomyelitis, subperiosteal abscess, subdural empyema, and dural venous sinus thrombosis.
Key points and pitfalls
Maintain a high index of suspicion in patients with persistent or inadequately treated otitis media. Failure to recognize meningitis or intracranial extension is a major pitfall. Definitive management requires timely surgical drainage in addition to antibiotics.

- Published on
Emergency and Acute Medicine-MDMA Poisoning
Basics description
MDMA (3,4-methylenedioxymethamphetamine), commonly known as ecstasy, is a Schedule I illicit drug used recreationally at rave parties, dance clubs, and college campuses. Onset of effects typically occurs 15–30 minutes after ingestion, with a duration of 2–6 hours. Pills often contain contaminants such as caffeine, ephedrine, dextromethorphan, or ketamine. Related compounds include MDA, MDEA, MDBA, and PMA. MDMA has an amphetamine-like structure that increases catecholamine release and a mescaline-like ring that enhances serotonergic and dopaminergic activity.
Etiology
Toxicity results from deliberate or accidental ingestion of MDMA, often compounded by coingestants or adulterants.
Diagnosis signs and symptoms
Patients may present with altered mental status, severe sympathomimetic features, or hyperthermia. Neurologic findings include excitation, delirium, hallucinations, seizures, coma, and cerebral edema. Cardiovascular manifestations include early hypertension, late hypotension, palpitations, ventricular dysrhythmias, and ectopy. Pulmonary edema may occur. Metabolic complications include hyponatremia, hypoglycemia, and syndrome of inappropriate antidiuretic hormone secretion. Musculoskeletal findings include bruxism, rigidity, and restlessness, with rhabdomyolysis leading to renal failure. Hepatic injury ranges from hepatitis to jaundice, and hematologic complications include disseminated intravascular coagulation. Gastrointestinal symptoms include nausea, vomiting, diarrhea, and abdominal cramping. Other features include hyperthermia, mydriasis, and nystagmus.
Essential workup and diagnostic testing
Diagnosis is primarily clinical, supported by history and examination. Core temperature measurement is critical. Laboratory evaluation includes electrolytes, glucose, BUN, creatinine, coagulation studies, creatine phosphokinase, liver function tests, and urine studies for myoglobin. Urine toxicology screening may show amphetamines but is unreliable for MDMA specificity. ECG commonly shows sinus tachycardia with possible dysrhythmias. Imaging is guided by presentation, with chest radiography for aspiration or pulmonary edema and head CT for suspected intracranial pathology.
Differential diagnosis
Cocaine or amphetamine overdose, anticholinergic toxicity, synthetic cathinone intoxication, serotonin syndrome, sepsis, thyroid storm, pheochromocytoma, or occult head injury.
Treatment and emergency management
Management is supportive and focused on airway protection, oxygenation, IV access, and continuous monitoring. Benzodiazepines are first-line for agitation, anxiety, and seizures. Aggressive IV hydration with isotonic fluids is essential, particularly in rhabdomyolysis. Hyperthermia is treated with active cooling and sedation. Hypertension may require agents such as nitroprusside or phentolamine, while hypotension is treated with fluids and vasopressors as needed. Severe rhabdomyolysis or renal failure may require hemodialysis. Patients should be monitored for at least 6 hours.
Disposition and follow-up
Admission is required for patients with altered mental status, seizures, cardiovascular instability, rhabdomyolysis, disseminated intravascular coagulation, or loss of behavioral control. Asymptomatic patients may be discharged after 6 hours of observation. Substance use counseling is recommended, with psychiatric evaluation for intentional overdose.
Key points and pitfalls
Always obtain a core temperature and evaluate for hyponatremia in patients with persistent altered mental status. Routine toxicology screens may not detect MDMA or coingestants. Consider non-toxicologic causes of deterioration and recognize that hyperthermia and rhabdomyolysis are major causes of morbidity and mortality.
- Published on
Emergency and Acute Medicine – Measles
Basics description
Measles, also known as rubeola, is a vaccine-preventable infectious disease that primarily affects children and is characterized by fever, cough, coryza, conjunctivitis, and an erythematous maculopapular rash. Due to widespread immunization, incidence is now low, but outbreaks continue to occur in nonimmunized or underimmunized populations.
Etiology
Measles is caused by the rubeola virus, a morbillivirus in the paramyxovirus family with negative-strand RNA. Humans are the only known reservoir. The virus is highly contagious and transmitted via respiratory droplets or direct contact; therefore, respiratory isolation must be initiated when measles is suspected. Outbreaks are most often associated with lapses in vaccination coverage.
Special populations
During pregnancy, measles infection increases the risk of spontaneous abortion and premature contractions but does not appear to cause congenital malformations. Pregnant patients should not receive MMR or MMRV vaccines. Adults born before 1957 are generally considered immune; however, health care workers should receive vaccination if serologic testing shows negative titers. In pediatrics, routine immunization with MMR or MMRV begins at ≥12 months of age, with a second dose at 4–6 years; catch-up doses must be spaced at least 4 weeks apart.
Diagnosis, signs, and symptoms
After an incubation period of approximately 10–12 days, patients develop a prodrome lasting 1–7 days with fever, malaise, cough, coryza, and conjunctivitis. Koplik spots—small white to grayish-blue lesions on the buccal mucosa—are pathognomonic and appear 1–2 days before the rash, disappearing shortly after rash onset. The classic maculopapular blanching rash develops 3–7 days into illness, beginning on the head and spreading downward. It may become confluent and occasionally petechial, rarely involving the palms and soles. Rash resolution occurs over several days and may be followed by desquamation.
Complications
Respiratory complications are common, with pneumonia occurring in approximately 6% of cases and representing the most frequent cause of death, particularly in immunocompromised patients. Otitis media, sinusitis, and diarrhea are also frequent. Neurologic complications include seizures and encephalitis, which may develop days after rash onset due to postinfectious autoimmune mechanisms. A rare but fatal late complication is subacute sclerosing panencephalitis, occurring years after infection. Cardiovascular complications such as myocarditis or conduction defects are uncommon but may be clinically significant in older adults.
Essential workup and diagnostic testing
Diagnosis is primarily clinical, based on the combination of fever, cough, coryza, conjunctivitis, and characteristic rash. Laboratory confirmation may include measles IgM and IgG serology or PCR for measles RNA. Viral isolation from blood, throat, nasopharynx, or urine is mainly for epidemiologic surveillance. CSF analysis is indicated if encephalitis is suspected, and chest radiography is obtained when pneumonia is a concern.
Differential diagnosis
Conditions to consider include rubella, scarlet fever, infectious mononucleosis, roseola, erythema infectiosum, enteroviral infections, Kawasaki disease, secondary syphilis, toxic shock syndrome, and drug reactions.
Treatment and emergency management
Management is primarily supportive, focusing on antipyretics, hydration, and monitoring for complications. Strict isolation is required. Postexposure prophylaxis for susceptible individuals includes MMR vaccination within 72 hours of exposure or intramuscular immune globulin within 6 days for high-risk contacts such as infants, pregnant patients, and immunocompromised individuals. Oxygenation and airway protection are essential in cases complicated by pneumonia or encephalitis.
Medications
The World Health Organization recommends vitamin A supplementation for children with measles, particularly in areas of deficiency, as it reduces morbidity and mortality. Dosing is age-based and administered once daily for two days.
Disposition and follow-up
Hospital admission is indicated for patients with severe pneumonia, dehydration, encephalitis, SSPE, immunocompromise, or significant comorbidities. Patients are contagious from 4 days before symptom onset until 4 days after rash appearance; immunocompromised individuals may remain contagious longer. Discharge is appropriate for stable patients without complications, with clear instructions on isolation and follow-up.
Pearls and pitfalls
Measles is one of the most contagious infectious diseases, with significant morbidity and mortality despite modern care. Early recognition and respiratory isolation are critical in health care settings. Immunocompromised patients may not develop the classic rash, increasing the risk of missed diagnosis. Vaccination remains the most effective preventive measure.
- Published on
Emergency and Acute Medicine – Meningitis
Basics description
Meningitis is an infection of the central nervous system characterized by inflammation of the leptomeninges and an increased white blood cell count in the cerebrospinal fluid (CSF). It commonly presents with fever, headache, neck stiffness, and altered mental status, although manifestations vary by age and immune status.
Etiology
Meningitis may be bacterial, viral, fungal, or noninfectious in origin. In neonates, common bacterial causes include group B Streptococcus, Escherichia coli, other enteric bacilli, and Listeria monocytogenes. In children and adults, Streptococcus pneumoniae and Neisseria meningitidis predominate, while elderly patients and those with alcoholism are at increased risk for pneumococcal infection, gram-negative bacilli, and Listeria. Neurosurgical patients are more likely to develop infections due to staphylococcal or gram-negative organisms. Immunocompromised patients, including those with AIDS, may develop tuberculosis, fungal, or syphilitic meningitis in addition to typical pathogens. Viral meningitis is common and often less severe, while chemical, drug-induced, or toxin-related meningitis is rare.
Clinical presentation
Typical symptoms include fever, headache, photophobia, and neck stiffness. Kernig and Brudzinski signs may be present but are neither sensitive nor specific. Patients may also develop altered mental status, seizures, focal neurologic deficits, papilledema, or a petechial or purpuric rash suggestive of meningococcal disease. Associated infections such as sinusitis, otitis media, or pneumonia may provide clues to the source.
Infants and young children often present atypically, with fever or hypothermia, lethargy, poor feeding, vomiting, respiratory distress, apnea, cyanosis, hypotonia, or a bulging fontanelle. Classic meningeal signs are frequently absent in children under one year of age. Elderly or immunocompromised patients may present primarily with confusion and fewer classic symptoms.
Essential workup
Management should begin immediately when meningitis is suspected. Blood cultures should be obtained promptly, followed by empiric antimicrobial therapy without delay, especially if the patient is unstable. Lumbar puncture is recommended for all patients with suspected meningitis unless contraindicated. Neuroimaging prior to lumbar puncture is reserved for patients at risk for herniation, including those with immunodeficiency, recent seizures, focal neurologic deficits, altered consciousness, papilledema, a history of central nervous system disease, or age over 60 years.
CSF analysis includes cell count and differential, glucose, protein, Gram stain, and culture, with additional studies added as clinically indicated. Elevated opening pressure may be present. Typical bacterial meningitis is associated with low CSF glucose, elevated protein, and a high white blood cell count with neutrophil predominance, although early disease may show fewer abnormalities.
Diagnostic tests and interpretation
Laboratory evaluation includes blood cultures, complete blood count, electrolytes, renal function tests, coagulation studies, and serum glucose for comparison with CSF glucose. Imaging such as head CT is used selectively to assess for contraindications to lumbar puncture or complications. Chest radiography may identify pneumonia or tuberculosis when suspected. Polymerase chain reaction testing and antigen detection may assist in identifying viral or bacterial pathogens when cultures are nondiagnostic.
Differential diagnosis
Conditions that may mimic meningitis include encephalitis, intracranial abscess, epidural or spinal abscess, febrile seizures, intracranial hemorrhage, stroke, systemic or central nervous system lupus, malignancy, venous sinus thrombosis, trauma, and metabolic or toxic encephalopathies.
Treatment and emergency management
Initial management focuses on airway, breathing, and circulation, seizure control, and isolation when appropriate. Empiric intravenous antibiotics should be administered as soon as possible, ideally after blood cultures but without delaying treatment for imaging or lumbar puncture. Antimicrobial regimens are selected based on patient age, immune status, and risk factors, with coverage for likely organisms. Vancomycin is added when resistant pneumococcal infection is a concern, and ampicillin is included in older adults and immunocompromised patients for Listeria coverage. Acyclovir should be started when herpes simplex encephalitis is suspected.
Adjunctive corticosteroids may reduce neurologic complications in selected patients, particularly those with pneumococcal meningitis, and should be given before or with the first dose of antibiotics when indicated.
Disposition and follow-up
Hospital admission is required for all patients with known or suspected bacterial meningitis, immunocompromised patients, and any toxic-appearing individual. Discharge from the emergency department is reserved for carefully selected patients with confirmed viral meningitis, stable clinical status, reliable follow-up, and clear discharge instructions coordinated with primary care.
Pearls and pitfalls
Meningitis rarely presents as a simple febrile seizure in children. Delayed recognition or treatment can lead to severe neurologic injury or death, making early empiric therapy essential. When clinical suspicion is high, treatment should never be postponed while awaiting diagnostic confirmation.
- Published on
KembaraXtra-Medicine – Atelectasis
Atelectasis refers to the collapse of part or all of the lung, resulting in loss of lung volume. It is broadly classified into two major types. Obstructive atelectasis, the most common form, occurs when an airway is blocked, such as by a mucus plug, foreign body, or tumor, leading to resorption of air in the alveoli beyond the obstruction. Nonobstructive atelectasis occurs without airway blockage and may result from external compression (for example, pleural effusion), surfactant abnormalities, lung scarring, or reduced ventilation of part of the lung.
Atelectasis is frequently seen in postoperative patients, individuals with lung or chest wall injury, and those receiving mechanical ventilation. Dependent regions of the lung are particularly prone to collapse. Prior asbestos exposure is associated with a characteristic form known as rounded atelectasis. There is no clear racial or sex predilection.
Clinical presentation varies widely. Some patients are asymptomatic, while others develop cough, dyspnea, and hypoxemia. On physical examination, findings over the affected area may include decreased or absent breath sounds, dullness to percussion, reduced tactile fremitus, and diminished vocal resonance.
The causes of atelectasis are diverse and include airway obstruction (such as tumors, mucus plugs, or foreign bodies), extrinsic bronchial compression from masses or enlarged cardiac structures, pleural disease (including pleural effusion, pneumothorax, mesothelioma, or rounded atelectasis), and alveolar injury from aspiration, toxic inhalation, infection, or acute respiratory distress syndrome. Additional contributors include chest wall abnormalities, impaired respiratory mechanics due to pain or neuromuscular disease, and repetitive shear injury during positive-pressure ventilation.
The differential diagnosis includes bronchogenic carcinoma, pneumonia, pulmonary infarction, and pleural effusion. Diagnostic evaluation typically begins with a chest x-ray, which often suggests volume loss. Thoracic ultrasonography can help distinguish atelectasis from pleural effusion or consolidation. Chest CT scanning is useful when endobronchial obstruction or external compression is suspected, and prone positioning may help differentiate dependent atelectasis from true consolidation. Bronchoscopy is reserved for selected cases to evaluate or relieve airway obstruction.
Management focuses on re-expanding the lung and addressing the underlying cause. Nonpharmacologic measures include deep breathing exercises, early mobilization and ambulation, incentive spirometry, and airway clearance techniques such as positive expiratory pressure devices, cough-assist devices, and chest physiotherapy. In selected patients, tracheal suctioning or bronchoscopy may be required to remove mucus plugs or foreign bodies.
Acute management may include supplemental oxygen, positive-pressure ventilation (such as CPAP or PEEP), mucolytic therapy for mucus plugging, bronchodilators when indicated, and adequate pain control, especially in postoperative or trauma patients to allow effective ventilation. Large pleural effusions, hemothorax, or empyema should be drained when contributing to lung collapse.
Chronic management depends on the cause and may involve ongoing airway clearance therapies, treatment of underlying lung disease, debulking or stenting of obstructing endobronchial lesions, or removal of sources of external compression such as effusions or masses. Prognosis varies and is largely determined by the underlying etiology and the patient’s overall clinical condition.
Patients benefit from education emphasizing frequent position changes, sitting upright when possible, early mobilization, and adherence to breathing exercises. Referral to pulmonology or interventional pulmonology is appropriate when bronchoscopy, tumor management, or specialized airway interventions are required.
Atelectasis refers to the collapse of part or all of the lung, resulting in loss of lung volume. It is broadly classified into two major types. Obstructive atelectasis, the most common form, occurs when an airway is blocked, such as by a mucus plug, foreign body, or tumor, leading to resorption of air in the alveoli beyond the obstruction. Nonobstructive atelectasis occurs without airway blockage and may result from external compression (for example, pleural effusion), surfactant abnormalities, lung scarring, or reduced ventilation of part of the lung.
Atelectasis is frequently seen in postoperative patients, individuals with lung or chest wall injury, and those receiving mechanical ventilation. Dependent regions of the lung are particularly prone to collapse. Prior asbestos exposure is associated with a characteristic form known as rounded atelectasis. There is no clear racial or sex predilection.
Clinical presentation varies widely. Some patients are asymptomatic, while others develop cough, dyspnea, and hypoxemia. On physical examination, findings over the affected area may include decreased or absent breath sounds, dullness to percussion, reduced tactile fremitus, and diminished vocal resonance.
The causes of atelectasis are diverse and include airway obstruction (such as tumors, mucus plugs, or foreign bodies), extrinsic bronchial compression from masses or enlarged cardiac structures, pleural disease (including pleural effusion, pneumothorax, mesothelioma, or rounded atelectasis), and alveolar injury from aspiration, toxic inhalation, infection, or acute respiratory distress syndrome. Additional contributors include chest wall abnormalities, impaired respiratory mechanics due to pain or neuromuscular disease, and repetitive shear injury during positive-pressure ventilation.
The differential diagnosis includes bronchogenic carcinoma, pneumonia, pulmonary infarction, and pleural effusion. Diagnostic evaluation typically begins with a chest x-ray, which often suggests volume loss. Thoracic ultrasonography can help distinguish atelectasis from pleural effusion or consolidation. Chest CT scanning is useful when endobronchial obstruction or external compression is suspected, and prone positioning may help differentiate dependent atelectasis from true consolidation. Bronchoscopy is reserved for selected cases to evaluate or relieve airway obstruction.
Management focuses on re-expanding the lung and addressing the underlying cause. Nonpharmacologic measures include deep breathing exercises, early mobilization and ambulation, incentive spirometry, and airway clearance techniques such as positive expiratory pressure devices, cough-assist devices, and chest physiotherapy. In selected patients, tracheal suctioning or bronchoscopy may be required to remove mucus plugs or foreign bodies.
Acute management may include supplemental oxygen, positive-pressure ventilation (such as CPAP or PEEP), mucolytic therapy for mucus plugging, bronchodilators when indicated, and adequate pain control, especially in postoperative or trauma patients to allow effective ventilation. Large pleural effusions, hemothorax, or empyema should be drained when contributing to lung collapse.
Chronic management depends on the cause and may involve ongoing airway clearance therapies, treatment of underlying lung disease, debulking or stenting of obstructing endobronchial lesions, or removal of sources of external compression such as effusions or masses. Prognosis varies and is largely determined by the underlying etiology and the patient’s overall clinical condition.
Patients benefit from education emphasizing frequent position changes, sitting upright when possible, early mobilization, and adherence to breathing exercises. Referral to pulmonology or interventional pulmonology is appropriate when bronchoscopy, tumor management, or specialized airway interventions are required.
- Published on
KembaraXtra-Medicine – Ataxia Telangiectasia (AT)
Ataxia telangiectasia (AT) is a rare autosomal recessive childhood disorder caused by defective DNA repair. It is a multisystem condition characterized by progressive cerebellar ataxia, abnormal movements (e.g., choreoathetosis), oculocutaneous telangiectasias, recurrent infections from immune dysfunction, marked sensitivity to ionizing radiation, and an increased risk of malignancy. The ICD-10CM code is G11.3 (cerebellar ataxia with defective DNA repair).
AT occurs in about 1 in 40,000 live births, affects males and females equally, and typically begins in early childhood (often ages 1–5 years). It is considered the second most common autosomal recessive ataxia after Friedreich ataxia. The causative gene is ATM (mapped to chromosome 11q22–23), and many different mutations have been identified. The ATM protein is a kinase activated by double-stranded DNA breaks, triggering pathways for cell cycle arrest, DNA repair, or apoptosis. ATM is also important for V(D)J recombination in developing lymphocytes, which helps explain the immune problems seen in AT.
Children often develop normally until they begin walking, when gait and truncal ataxia become noticeable. Over time, symptoms can include slurred speech, progressive difficulty coordinating eye movements (including oculomotor apraxia), polyneuropathy, choreoathetosis, delayed physical and sexual development, and features of premature aging (such as early graying and skin/subcutaneous fat changes). Motor skills gradually decline, and by the second decade, many individuals require a wheelchair for at least part of the day. As the condition progresses, oromotor and swallowing difficulties can increase the risk of aspiration. Cognitive function is often initially normal and may decline early, then tends to plateau around about 10 years of age, though motor impairment can make school performance and timed testing look worse than true cognitive ability.
Telangiectasias are highly suggestive of AT but often appear later—typically after age 6—which can delay diagnosis. They commonly involve the bulbar conjunctiva (sometimes mistaken for conjunctivitis) and can also appear on the ears, cheeks, corners of the eyes, neck, nasal bridge, and flexor forearm creases. Skin findings may include café-au-lait macules, hypopigmented macules, or melanocytic nevi. Immune dysfunction affects 60%–80% of patients and stems from B-cell and T-helper problems. IgA is absent in many patients, IgE is low/absent in most, and IgM may be elevated; this predisposes to recurrent sinopulmonary infections. While the immune deficiency itself is typically not progressive, repeated infections and aspiration can contribute to bronchiectasis later in life.
Neuropathology includes progressive cerebellar injury with loss of Purkinje cells, and later changes may involve posterior columns and spinocerebellar tracts, with reports of anterior horn cell involvement and peripheral nerve changes. Cancer risk becomes increasingly important with age: beyond about 10 years, the incidence has been described as roughly 1% per year, with an overall lifetime risk around 30%–40%, most commonly leukemia and lymphoma. Carriers (heterozygotes) usually do not have classic AT symptoms but may have a higher malignancy risk at younger ages.
Diagnosis is based on the clinical picture (early-onset progressive ataxia plus telangiectasia and/or immune abnormalities), family history, and supportive testing. The differential for early-onset ataxia includes Friedreich ataxia, abetalipoproteinemia, acquired vitamin E deficiency, AOA1/AOA2, AT-like disorder, various spinocerebellar ataxias, certain metabolic diseases (e.g., Hartnup disease, maple syrup urine disease), and leukodystrophies. Helpful laboratory findings can include abnormal immunoglobulin levels, and serum α-fetoprotein, which is elevated in more than 95% of children with AT older than about 8 months. Some patients have acanthocytes on smear. Immunoblot testing for ATM protein is highly sensitive/specific, and many affected individuals have little to no detectable ATM protein. Brain MRI or CT typically shows cerebellar atrophy during childhood; imaging may also show findings compatible with telangiectasia-related changes.
There is no proven disease-specific cure, so management focuses on monitoring and reducing complications. This includes close surveillance for infections and cancers, consideration of IV immunoglobulin for those with frequent/severe infections, and sometimes prophylactic antibiotics. Routine preventive care is important, including influenza and pneumococcal vaccination. Because of radiation sensitivity, exposure to ionizing radiation should be minimized, including limiting diagnostic radiographs when possible. Swallowing should be monitored as needed to reduce aspiration risk, and physical/occupational therapy supports mobility and helps prevent contractures. Some studies suggest possible symptomatic improvement with glucocorticoids, thought to influence ATM splicing and preserve some kinase activity, but this is not established as definitive therapy. Antioxidants such as vitamin E are sometimes used empirically, though strong evidence is limited.
Prognosis is serious due to multisystem involvement, but survival has improved; many individuals now live beyond 25 years, and some into their 40s or 50s. Morbidity and mortality are most often related to recurrent infections or malignancy. Ongoing care typically involves coordinated referral to immunology, neurology, gastroenterology/nutrition, rehabilitation services, and genetic counseling.
Ataxia telangiectasia (AT) is a rare autosomal recessive childhood disorder caused by defective DNA repair. It is a multisystem condition characterized by progressive cerebellar ataxia, abnormal movements (e.g., choreoathetosis), oculocutaneous telangiectasias, recurrent infections from immune dysfunction, marked sensitivity to ionizing radiation, and an increased risk of malignancy. The ICD-10CM code is G11.3 (cerebellar ataxia with defective DNA repair).
AT occurs in about 1 in 40,000 live births, affects males and females equally, and typically begins in early childhood (often ages 1–5 years). It is considered the second most common autosomal recessive ataxia after Friedreich ataxia. The causative gene is ATM (mapped to chromosome 11q22–23), and many different mutations have been identified. The ATM protein is a kinase activated by double-stranded DNA breaks, triggering pathways for cell cycle arrest, DNA repair, or apoptosis. ATM is also important for V(D)J recombination in developing lymphocytes, which helps explain the immune problems seen in AT.
Children often develop normally until they begin walking, when gait and truncal ataxia become noticeable. Over time, symptoms can include slurred speech, progressive difficulty coordinating eye movements (including oculomotor apraxia), polyneuropathy, choreoathetosis, delayed physical and sexual development, and features of premature aging (such as early graying and skin/subcutaneous fat changes). Motor skills gradually decline, and by the second decade, many individuals require a wheelchair for at least part of the day. As the condition progresses, oromotor and swallowing difficulties can increase the risk of aspiration. Cognitive function is often initially normal and may decline early, then tends to plateau around about 10 years of age, though motor impairment can make school performance and timed testing look worse than true cognitive ability.
Telangiectasias are highly suggestive of AT but often appear later—typically after age 6—which can delay diagnosis. They commonly involve the bulbar conjunctiva (sometimes mistaken for conjunctivitis) and can also appear on the ears, cheeks, corners of the eyes, neck, nasal bridge, and flexor forearm creases. Skin findings may include café-au-lait macules, hypopigmented macules, or melanocytic nevi. Immune dysfunction affects 60%–80% of patients and stems from B-cell and T-helper problems. IgA is absent in many patients, IgE is low/absent in most, and IgM may be elevated; this predisposes to recurrent sinopulmonary infections. While the immune deficiency itself is typically not progressive, repeated infections and aspiration can contribute to bronchiectasis later in life.
Neuropathology includes progressive cerebellar injury with loss of Purkinje cells, and later changes may involve posterior columns and spinocerebellar tracts, with reports of anterior horn cell involvement and peripheral nerve changes. Cancer risk becomes increasingly important with age: beyond about 10 years, the incidence has been described as roughly 1% per year, with an overall lifetime risk around 30%–40%, most commonly leukemia and lymphoma. Carriers (heterozygotes) usually do not have classic AT symptoms but may have a higher malignancy risk at younger ages.
Diagnosis is based on the clinical picture (early-onset progressive ataxia plus telangiectasia and/or immune abnormalities), family history, and supportive testing. The differential for early-onset ataxia includes Friedreich ataxia, abetalipoproteinemia, acquired vitamin E deficiency, AOA1/AOA2, AT-like disorder, various spinocerebellar ataxias, certain metabolic diseases (e.g., Hartnup disease, maple syrup urine disease), and leukodystrophies. Helpful laboratory findings can include abnormal immunoglobulin levels, and serum α-fetoprotein, which is elevated in more than 95% of children with AT older than about 8 months. Some patients have acanthocytes on smear. Immunoblot testing for ATM protein is highly sensitive/specific, and many affected individuals have little to no detectable ATM protein. Brain MRI or CT typically shows cerebellar atrophy during childhood; imaging may also show findings compatible with telangiectasia-related changes.
There is no proven disease-specific cure, so management focuses on monitoring and reducing complications. This includes close surveillance for infections and cancers, consideration of IV immunoglobulin for those with frequent/severe infections, and sometimes prophylactic antibiotics. Routine preventive care is important, including influenza and pneumococcal vaccination. Because of radiation sensitivity, exposure to ionizing radiation should be minimized, including limiting diagnostic radiographs when possible. Swallowing should be monitored as needed to reduce aspiration risk, and physical/occupational therapy supports mobility and helps prevent contractures. Some studies suggest possible symptomatic improvement with glucocorticoids, thought to influence ATM splicing and preserve some kinase activity, but this is not established as definitive therapy. Antioxidants such as vitamin E are sometimes used empirically, though strong evidence is limited.
Prognosis is serious due to multisystem involvement, but survival has improved; many individuals now live beyond 25 years, and some into their 40s or 50s. Morbidity and mortality are most often related to recurrent infections or malignancy. Ongoing care typically involves coordinated referral to immunology, neurology, gastroenterology/nutrition, rehabilitation services, and genetic counseling.
- Published on
KembaraXtra-Medicine – Ataxia
Ataxia most commonly refers to an unsteady, uncoordinated gait, but the term is also used more broadly to describe incoordination that can involve the limbs, trunk, speech and swallowing muscles, and even eye movements. Many people first notice imbalance while walking, frequent falls, or “bumping into” walls. On exam, gait ataxia often appears as a broad-based, unsteady gait with difficulty performing tandem walking (heel-to-toe).
Ataxia can affect all ages and both sexes equally. Hereditary ataxias are estimated to affect about 1 in 50,000 people in the United States, and overall prevalence has been reported around 13.9 per 10,000. Hereditary forms often peak in young adulthood, while new ataxia after age 65 is commonly related to stroke. Risk factors include a family history of ataxia, heavy alcohol use, hypothyroidism, chronic anticonvulsant use (especially phenytoin), celiac disease, and cerebrovascular disease. The most common inherited ataxia in the U.S. is Friedreich ataxia (autosomal recessive), followed by autosomal dominant spinocerebellar ataxias, with SCA3 being a common subtype.
Symptoms typically progress from mild unsteadiness to frequent falls. People may also report double vision or a sensation that the environment is moving, and speech can become slurred and harder to understand. Physical findings may include nystagmus and other abnormal eye movements, slurred speech, dysmetria (past-pointing) on finger-to-nose or heel-to-shin testing, and dysdiadochokinesia (difficulty with rapid alternating movements). Gait is often wide-based with trouble turning and a tendency to veer or sway, and tandem gait is usually abnormal.
Causes of ataxia are diverse and are often grouped into cerebellar ataxia (from cerebellar dysfunction or its connections) and sensory ataxia (from impaired proprioception due to dorsal column disease, dorsal root ganglionopathy, or large-fiber neuropathy). Etiologies include genetic disorders (e.g., Friedreich ataxia, spinocerebellar ataxias, ataxia-telangiectasia, episodic ataxias), vascular events (cerebellar stroke), infections and inflammatory conditions (postinfectious cerebellitis, multiple sclerosis, HIV, Whipple disease), tumors, structural abnormalities (Arnold–Chiari malformation), toxins (alcohol, certain chemotherapies, heavy metals, solvents), medications (especially phenytoin), endocrine disease (hypothyroidism), and nutritional deficiencies (vitamin E, B1, B12). Autoimmune and paraneoplastic syndromes are also important considerations, including gluten ataxia, Hashimoto encephalopathy, anti-GAD65 ataxia, and subacute cerebellar degeneration associated with antibodies such as anti-Yo.
Evaluation should be systematic and prioritize reversible causes. A careful family history can guide genetic testing, and acute-onset ataxia should prompt urgent assessment for stroke with appropriate brain imaging. Brain MRI (with and without contrast when appropriate) is strongly recommended to assess for infarct, hemorrhage, tumor, inflammation, or cerebellar atrophy; CT is used when MRI is not feasible. Lumbar puncture may be needed if infection, inflammatory disease, or leptomeningeal malignancy is suspected. When cancer is suspected or known, a paraneoplastic antibody panel may be appropriate, recognizing that neurologic paraneoplastic syndromes can precede detectable malignancy.
Common baseline laboratory testing often includes a complete blood count, comprehensive metabolic panel and liver tests, thyroid function tests, and screening for infections such as HIV and syphilis (RPR). Nutritional and metabolic evaluation may include folate, vitamin B12 (with methylmalonic acid/homocysteine when B12 is “low-normal”), vitamin E (interpreted with cholesterol), and tests for celiac disease (e.g., tissue transglutaminase antibodies). Copper and ceruloplasmin may be checked when Wilson disease is a concern. Electromyography and nerve conduction studies are useful when sensory ataxia due to large-fiber neuropathy or myeloneuropathy is suspected. Selected cases may require genetic testing, autoimmune markers (e.g., anti-GAD65, anti-thyroid antibodies), or specialized metabolic testing depending on age of onset and clinical pattern.
Treatment depends on the underlying cause whenever possible. Reversible contributors are addressed directly—for example, vitamin replacement for deficiencies, treating hypothyroidism, stopping or adjusting offending toxins/medications, and disease-specific therapy for immune or inflammatory causes when indicated. Sudden ataxia must be evaluated and treated as possible stroke using standard acute stroke pathways. For many genetic or degenerative ataxias, care is largely supportive and focused on preventing complications and maintaining function, including monitoring for associated problems such as neuropathy or cardiac disease in certain inherited forms.
Rehabilitation is central for nearly all patients with ataxia. Physical therapy can improve gait and balance, occupational therapy can support daily function and safety, and speech therapy can help with dysarthria and swallowing issues. Long-term outlook varies: reversible causes can improve significantly with treatment, while many inherited ataxias are progressive. Neurology referral is recommended for diagnostic guidance, genetic evaluation when appropriate, and ongoing management. Families should be educated on fall prevention and home safety measures to reduce injury risk, and a gluten-free diet is recommended when ataxia is related to celiac disease.
Ataxia most commonly refers to an unsteady, uncoordinated gait, but the term is also used more broadly to describe incoordination that can involve the limbs, trunk, speech and swallowing muscles, and even eye movements. Many people first notice imbalance while walking, frequent falls, or “bumping into” walls. On exam, gait ataxia often appears as a broad-based, unsteady gait with difficulty performing tandem walking (heel-to-toe).
Ataxia can affect all ages and both sexes equally. Hereditary ataxias are estimated to affect about 1 in 50,000 people in the United States, and overall prevalence has been reported around 13.9 per 10,000. Hereditary forms often peak in young adulthood, while new ataxia after age 65 is commonly related to stroke. Risk factors include a family history of ataxia, heavy alcohol use, hypothyroidism, chronic anticonvulsant use (especially phenytoin), celiac disease, and cerebrovascular disease. The most common inherited ataxia in the U.S. is Friedreich ataxia (autosomal recessive), followed by autosomal dominant spinocerebellar ataxias, with SCA3 being a common subtype.
Symptoms typically progress from mild unsteadiness to frequent falls. People may also report double vision or a sensation that the environment is moving, and speech can become slurred and harder to understand. Physical findings may include nystagmus and other abnormal eye movements, slurred speech, dysmetria (past-pointing) on finger-to-nose or heel-to-shin testing, and dysdiadochokinesia (difficulty with rapid alternating movements). Gait is often wide-based with trouble turning and a tendency to veer or sway, and tandem gait is usually abnormal.
Causes of ataxia are diverse and are often grouped into cerebellar ataxia (from cerebellar dysfunction or its connections) and sensory ataxia (from impaired proprioception due to dorsal column disease, dorsal root ganglionopathy, or large-fiber neuropathy). Etiologies include genetic disorders (e.g., Friedreich ataxia, spinocerebellar ataxias, ataxia-telangiectasia, episodic ataxias), vascular events (cerebellar stroke), infections and inflammatory conditions (postinfectious cerebellitis, multiple sclerosis, HIV, Whipple disease), tumors, structural abnormalities (Arnold–Chiari malformation), toxins (alcohol, certain chemotherapies, heavy metals, solvents), medications (especially phenytoin), endocrine disease (hypothyroidism), and nutritional deficiencies (vitamin E, B1, B12). Autoimmune and paraneoplastic syndromes are also important considerations, including gluten ataxia, Hashimoto encephalopathy, anti-GAD65 ataxia, and subacute cerebellar degeneration associated with antibodies such as anti-Yo.
Evaluation should be systematic and prioritize reversible causes. A careful family history can guide genetic testing, and acute-onset ataxia should prompt urgent assessment for stroke with appropriate brain imaging. Brain MRI (with and without contrast when appropriate) is strongly recommended to assess for infarct, hemorrhage, tumor, inflammation, or cerebellar atrophy; CT is used when MRI is not feasible. Lumbar puncture may be needed if infection, inflammatory disease, or leptomeningeal malignancy is suspected. When cancer is suspected or known, a paraneoplastic antibody panel may be appropriate, recognizing that neurologic paraneoplastic syndromes can precede detectable malignancy.
Common baseline laboratory testing often includes a complete blood count, comprehensive metabolic panel and liver tests, thyroid function tests, and screening for infections such as HIV and syphilis (RPR). Nutritional and metabolic evaluation may include folate, vitamin B12 (with methylmalonic acid/homocysteine when B12 is “low-normal”), vitamin E (interpreted with cholesterol), and tests for celiac disease (e.g., tissue transglutaminase antibodies). Copper and ceruloplasmin may be checked when Wilson disease is a concern. Electromyography and nerve conduction studies are useful when sensory ataxia due to large-fiber neuropathy or myeloneuropathy is suspected. Selected cases may require genetic testing, autoimmune markers (e.g., anti-GAD65, anti-thyroid antibodies), or specialized metabolic testing depending on age of onset and clinical pattern.
Treatment depends on the underlying cause whenever possible. Reversible contributors are addressed directly—for example, vitamin replacement for deficiencies, treating hypothyroidism, stopping or adjusting offending toxins/medications, and disease-specific therapy for immune or inflammatory causes when indicated. Sudden ataxia must be evaluated and treated as possible stroke using standard acute stroke pathways. For many genetic or degenerative ataxias, care is largely supportive and focused on preventing complications and maintaining function, including monitoring for associated problems such as neuropathy or cardiac disease in certain inherited forms.
Rehabilitation is central for nearly all patients with ataxia. Physical therapy can improve gait and balance, occupational therapy can support daily function and safety, and speech therapy can help with dysarthria and swallowing issues. Long-term outlook varies: reversible causes can improve significantly with treatment, while many inherited ataxias are progressive. Neurology referral is recommended for diagnostic guidance, genetic evaluation when appropriate, and ongoing management. Families should be educated on fall prevention and home safety measures to reduce injury risk, and a gluten-free diet is recommended when ataxia is related to celiac disease.