- Published on
KembaraXtra- Medicine – Adrenal Insufficiency
Adrenal insufficiency refers to inadequate production of adrenal corticosteroids. It most classically results from partial or complete destruction of the adrenal cortex causing primary adrenal failure (Addison disease), in which cortisol deficiency is typically accompanied by impaired mineralocorticoid (aldosterone) production. In contrast, secondary cortisol deficiency results from pituitary (or hypothalamic) dysfunction leading to reduced ACTH stimulation of the adrenal glands, causing low cortisol but usually preserved aldosterone secretion, since aldosterone is mainly regulated by the renin–angiotensin system. Adrenal insufficiency can present gradually with nonspecific symptoms, and a high index of suspicion is required because up to about half of patients may first present with an acute adrenal crisis.
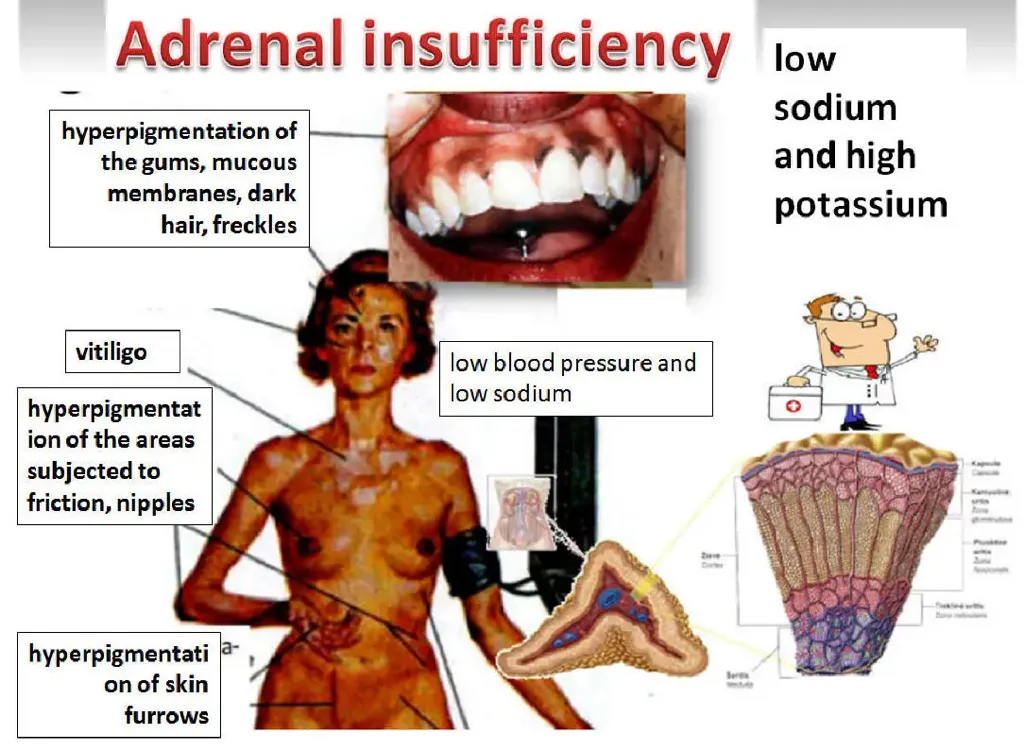
Adrenal insufficiency is relatively uncommon, with a prevalence of 10–15 per 100,000 persons, and occurs more often in females than males with an approximately 2:1 ratio. Clinical presentation may be insidious and includes generalized weakness, chronic fatigue, malaise, anorexia, weight loss, nausea, vomiting, abdominal discomfort, and postural dizziness. Hyperpigmentation of the skin and mucous membranes is a hallmark of primary adrenal insufficiency due to elevated ACTH and related melanocyte-stimulating activity; it is most prominent in palmar creases, buccal mucosa, pressure points (elbows, knees, knuckles), perianal mucosa, scars, and around the areolae. Hypotension and orthostasis are common. Women may have amenorrhea and loss of axillary hair due to adrenal androgen deficiency. Laboratory abnormalities often include hyponatremia, hyperkalemia (mainly in primary disease), hypoglycemia, and prerenal azotemia (elevated BUN/creatinine ratio). Additional findings can include mild normocytic anemia, neutropenia, lymphocytosis, eosinophilia, metabolic acidosis, and occasional hypercalcemia; severe dehydration can sometimes mask hyponatremia and anemia.
The most common cause of primary adrenal insufficiency is autoimmune adrenalitis, accounting for about 80% of cases. Other important causes include tuberculosis (historically significant, still relevant in some settings), metastatic or infiltrative malignancy (including lymphoma), adrenal hemorrhage (associated with anticoagulation, trauma, coagulopathies, pregnancy, or sepsis), adrenal infarction (e.g., antiphospholipid syndrome, thrombosis, arteritis), infections in immunocompromised states such as AIDS (with CMV adrenalitis), and genetic or syndromic causes including autoimmune polyglandular syndromes types 1 and 2, X-linked adrenoleukodystrophy, and congenital adrenal hyperplasia. Additional infiltrative etiologies include sarcoidosis, amyloidosis, hemochromatosis, vasculitides such as granulomatosis with polyangiitis, postoperative adrenal loss, and fungal infections such as histoplasmosis and candidiasis. Secondary adrenal insufficiency is usually due to pituitary disease, pituitary surgery or radiation, infiltrative pituitary disorders, or suppression of the hypothalamic–pituitary–adrenal axis following chronic exogenous glucocorticoid therapy.
Because symptoms overlap with many disorders, the differential diagnosis includes sepsis, hypovolemic shock, acute abdomen, depression, anorexia nervosa, chronic infection, gastrointestinal malignancy, myopathies, salt-losing nephritis, hemochromatosis, and in older adults “apathetic hyperthyroidism.” Diagnostic evaluation typically begins with assessment of cortisol production using an early morning (around 8 A.M.) serum cortisol. A cortisol level <3 mcg />L is strongly consistent with cortisol deficiency, while levels >18 mcg/dL generally exclude adrenal insufficiency. If the morning cortisol is 3-15 mcg/dL, confirmation is performed with a rapid ACTH (cosyntropin/Synacthen) stimulation test using 250 mcg ACTH IV, measuring cortisol at baseline, 30 minutes, and 60 minutes. A peak cortisol response >18 mcg/dL (≈500 nmol/L) suggests normal adrenal reserve, whereas failure to reach this threshold supports adrenal insufficiency. After confirming inadequate cortisol response, measuring plasma ACTH helps distinguish primary from secondary disease: ACTH >200 pg/mL supports primary adrenal insufficiency, while normal or low ACTH suggests secondary adrenal insufficiency. In critical illness–related corticosteroid insufficiency, a low-dose 1 mcg ACTH stimulation approach may be used, and an inadequate response may be defined by a cortisol level <25 mcg />L or an increment <9 mcg />L above baseline, depending on the clinical context.
Further evaluation aims to identify the cause. 21-hydroxylase antibodies are present in approximately 90% of autoimmune adrenalitis; if negative, adrenal imaging (typically CT) is recommended to assess for infection, hemorrhage, malignancy, or infiltrative disease. Tuberculosis evaluation may include PPD or interferon-gamma release testing. Imaging is not required to establish the diagnosis but can support etiology: small adrenal glands may indicate long-standing autoimmune atrophy or chronic tuberculosis, whereas enlarged glands can suggest early TB or other potentially treatable infiltrative processes. Chest radiography may show a small cardiac silhouette, and abdominal radiography can occasionally reveal adrenal calcifications in TB or fungal disease.
Management includes both chronic hormone replacement and urgent treatment of adrenal crisis. Patients should be educated on long-term monitoring of electrolytes, blood pressure, and weight, and generally advised to maintain a liberal sodium intake, especially in primary disease. They should carry a MedicAlert bracelet and an emergency kit containing injectable hydrocortisone, and both patients and partners should be trained to administer intramuscular steroids if vomiting or impaired consciousness prevents oral intake. Bone density monitoring may be considered because long-term glucocorticoid exposure increases osteoporosis risk.
Adrenal crisis is a life-threatening emergency characterized by circulatory collapse, dehydration, nausea/vomiting, hypoglycemia, hyperkalemia, and often severe hypotension. Treatment must begin immediately and should not be delayed for confirmatory testing. Initial steps include establishing large-bore IV access, drawing blood for electrolytes, glucose, cortisol, and ACTH, and rapidly infusing 2–3 liters of isotonic saline (0.9% NaCl) or dextrose-containing saline if hypoglycemia is present, with careful monitoring for fluid overload. Hydrocortisone 100 mg IV should be administered immediately and continued 100 mg every 6 hours, alongside supportive care and aggressive identification and treatment of precipitating causes such as infection, hemorrhage, or trauma. After stabilization, IV fluids are continued more slowly over 24–48 hours, the diagnosis is confirmed if previously unknown, glucocorticoids are tapered to maintenance dosing over 1–3 days as clinically appropriate, and mineralocorticoid replacement with fludrocortisone is started when stress-dose hydrocortisone is reduced and saline infusion is stopped.
Chronic treatment of primary adrenal insufficiency requires both glucocorticoid and mineralocorticoid replacement. A common regimen is hydrocortisone 15–20 mg on awakening with 5–10 mg in the early afternoon, or an alternative prednisone schedule such as 5 mg in the morning and 2.5 mg at bedtime, tailored to symptoms and avoidance of overtreatment. Fludrocortisone is generally required in primary disease, often starting around 0.1 mg daily (adjusted within a wider range based on orthostatic symptoms, blood pressure, edema, serum potassium, and plasma renin activity). Patients must be taught “sick day rules,” typically increasing glucocorticoid dosing two- to threefold for minor febrile illnesses for a few days without changing fludrocortisone dose, and using parenteral steroids if vomiting, diarrhea, severe illness, trauma, or surgery occurs. Stress-dose steroid coverage varies by severity: moderate illness may require hydrocortisone 50 mg twice daily orally or IV, while severe illness or shock can require hydrocortisone 100 mg IV every 8 hours with rapid tapering as recovery occurs. Minor procedures under local anesthesia often need no additional supplementation, whereas more stressful procedures may require a single 100 mg IV hydrocortisone dose just before the procedure, and major surgery typically requires 100 mg IV before induction followed by continued dosing for at least 24 hours with tapering.
In secondary adrenal insufficiency, mineralocorticoid replacement is usually unnecessary, and clinical clues include absence of hyperpigmentation and evidence of other pituitary hormone deficiencies such as hypothyroidism or hypogonadism. A critical safety point is that patients with both hypothyroidism and adrenal insufficiency should receive glucocorticoid replacement before thyroid hormone replacement, because thyroid hormone can increase cortisol clearance and precipitate adrenal crisis. Withdrawal from chronic exogenous steroid therapy should be gradual in patients treated for more than several weeks, with taper schedules individualized to dose and duration, and confirmation of hypothalamic–pituitary–adrenal axis recovery using appropriate stimulation testing when clinically indicated.
- Published on
KembaraXtra- Medicine – Adult-Onset Still Disease
Adult-onset Still disease (AOSD) is a rare systemic inflammatory disorder characterized by episodes of high-spiking fever, an evanescent rash, and arthralgias/arthritis, often accompanied by lymphadenopathy, hepatosplenomegaly, and markedly elevated serum ferritin. Many patients report a prodromal sore throat, sometimes preceding systemic symptoms. The daily (quotidian) fever pattern is a striking feature, and inflammatory arthritis can become progressive and destructive over time, leading to joint damage and disability in a subset of patients.
AOSD has several synonyms, including Still disease, systemic juvenile idiopathic arthritis (systemic JIA), and Wissler (Wissler-Fanconi) syndrome. The ICD-10-CM code is M06.1. Because it is uncommon, definitive epidemiologic data are limited, but prevalence has been reported between 1 and 34 per 1,000,000, with incidence around 0.1 to 0.4 per 100,000. A bimodal age distribution is described, with peaks around 15–25 and 36–46 years, and there is no consistent gender predisposition.
Infectious triggers are suspected in genetically susceptible individuals, as onset or relapse may follow a viral-like prodrome with pharyngitis, malaise, fever, and rash, although no single organism has been consistently implicated. Studies suggest a genetic component, with some associations reported between AOSD and certain HLA types (including class II DRB115 and DRB112 for susceptibility, and class I B35 with self-limited disease, DR2/DR5 with more chronic disease), though findings have not been consistently verified across populations.
Clinically, the “cardinal” triad of AOSD is fever, arthralgias/arthritis, and rash, and it occurs in most patients. Fever typically follows a quotidian pattern with spikes often occurring in the late afternoon or evening, frequently exceeding 102°F (38.9°C) and lasting a few hours before returning to normal. A “double quotidian” pattern with a second early-morning spike occurs in some cases. The classic rash is a salmon-pink, nonpruritic, maculopapular eruption most often involving the trunk and proximal extremities. The rash may appear with fever and fade when afebrile, which can make it easy to miss; Koebner phenomenon can occur. Arthralgias and myalgias are very common, and inflammatory arthritis often presents as symmetric polyarthritis affecting larger joints, with up to half of patients developing progressive destructive arthritis. Other manifestations may include sore throat, lymphadenopathy, hepatosplenomegaly, and serositis. Cardiac involvement such as pericarditis can occur and, in severe cases, can progress to tamponade, warranting prompt recognition and treatment.
The pathogenesis is not fully defined, but the disease appears driven by innate immune dysregulation and abnormal macrophage activation. Elevated proinflammatory cytokines are commonly described, including IL-1, IL-6, IL-18, TNF-α, interferon-γ, and others. More recently, increased circulating markers related to neutrophil extracellular traps (NETs) have been observed in active disease, with reductions after treatment, though the clinical implications continue to be studied.
There is no single diagnostic test that is pathognomonic for AOSD. Diagnosis is clinical and laboratory-based and is fundamentally a diagnosis of exclusion, requiring evaluation for infectious, neoplastic, and other inflammatory causes of fever and systemic illness. Laboratory abnormalities often include neutrophilic leukocytosis (commonly WBC >12 with >80% PMNs), very elevated inflammatory markers (often ESR >50 and CRP markedly elevated), normocytic anemia, thrombocytosis, hypoalbuminemia, and mild transaminitis. Creatine kinase (CPK) is typically normal even when myalgias are prominent. Hyperferritinemia is a particularly suggestive feature, and ferritin levels can exceed five times the upper limit of normal. Autoantibodies typical of other rheumatologic diseases are usually absent, with negative rheumatoid factor and negative ANA supporting (but not confirming) the diagnosis.
Imaging can support assessment of systemic involvement and complications. Plain radiographs may show early nonspecific changes such as soft-tissue swelling, effusions, or periarticular osteopenia, while later disease can demonstrate destructive arthropathy—classically in the wrists with joint-space narrowing and eventual ankylosis, and in hips with progressive joint destruction. CT or MRI may demonstrate lymphadenopathy, hepatosplenomegaly, and early bony or soft-tissue findings. FDG PET/CT may show increased uptake in spleen, bone marrow, and reactive lymph nodes, though these findings are not specific and must be interpreted in context.
Multiple classification/diagnostic criteria sets exist. The Yamaguchi criteria are widely used because they are sensitive and practical. The Fautrel criteria are more specific but require measurement of glycosylated ferritin, which is not routinely available in many settings. Regardless of criteria, careful exclusion of mimics is essential.
Prognosis is often favorable overall, with reported 5-year survival around 90%–95%, but many patients require long-term treatment to control inflammation and prevent relapse or progressive joint damage. Clinical course typically falls into one of three patterns: a self-limited/monophasic course with remission within a year, an intermittent/relapsing course with recurrent flares separated by remission, or a chronic/progressive course dominated by persistent arthritis and erosive destruction. Features associated with progression to chronic articular disease include polyarthritis, large-joint involvement, and very high ferritin levels at presentation.
Treatment depends on severity and organ involvement. Mild disease may respond to full-dose NSAIDs alone, though sustained control occurs in a minority. Most patients require systemic glucocorticoids, particularly when systemic symptoms are prominent or there is significant organ involvement. For steroid-sparing and control of arthritis, methotrexate is commonly used. Biologic therapy is frequently effective, especially for refractory disease or patients who cannot taper steroids. IL-1 blockade (such as anakinra or canakinumab) is often highly effective for systemic symptoms and inflammation. IL-6 inhibitors (such as tocilizumab or sarilumab) and TNF inhibitors (such as etanercept or infliximab) are also used, sometimes in combination with methotrexate. Response is typically reassessed after 2–3 months, and nonresponders are often switched to a biologic with a different mechanism. After prolonged remission, cautious tapering may be attempted. In severe or refractory cases, other therapies (including IVIG, and less commonly agents like rituximab or abatacept) have been reported with limited evidence.
A critical complication to recognize is macrophage activation syndrome (MAS), also referred to as reactive hemophagocytic lymphohistiocytosis. It occurs in a notable minority of AOSD patients and can be life-threatening, with substantial mortality. MAS can resemble a severe AOSD flare, but clues that raise suspicion include cytopenias (leukopenia and thrombocytopenia), hypofibrinogenemia, hypertriglyceridemia, and sometimes a paradoxical normalization of ESR despite clinical worsening. Bone marrow aspiration demonstrating hemophagocytosis is considered the diagnostic gold standard. Initial management commonly includes pulse-dose corticosteroids, IVIG, and cytokine-directed biologic therapy (often IL-1 or IL-6 blockade), alongside intensive supportive care.


- Published on
KembaraXtra- Medicine – Alcohol Use Disorder
Alcohol use disorder (AUD) describes problematic patterns of alcohol use that lead to clinically significant impairment or distress, and it includes a spectrum from hazardous use to dependence and withdrawal syndromes. “Moderate drinking” is commonly defined as up to two standard drinks per day for men and one standard drink per day for women and adults older than 65 years. “Hazardous” or “at-risk” drinking is defined as more than 14 drinks per week or more than 4 drinks per occasion for men, and 4 or more drinks on one occasion or 8 or more drinks per week for women. Alcohol withdrawal is defined by the development of characteristic symptoms after cessation or reduction of heavy and prolonged alcohol use, including autonomic hyperactivity, tremor, insomnia, nausea/vomiting, transient hallucinations or illusions, psychomotor agitation, anxiety, and grand mal seizures; symptoms must cause clinically significant distress or impairment and not be better explained by another condition.
AUD is also referred to as alcohol dependence syndrome, alcoholism, substance abuse, and alcohol withdrawal syndrome. ICD-10-CM codes fall under F10 (mental and behavioral disorders due to alcohol use), including F10.1 (harmful use), F10.2 (dependence syndrome), F10.3 (withdrawal state), F10.4 (withdrawal state with delirium), F10.5 (psychotic disorder), and F10.6 (amnesic syndrome). In U.S. clinical settings, alcohol-related problems are suggested by the clinical history in roughly 15% to 20% of primary care and hospitalized patients, with substantial economic and health impacts. AUD prevalence in adults (≥18 years) is commonly cited around 7%, with peak incidence in early adulthood and a common initial treatment age range in the mid-30s to mid-40s. Lifetime risk is higher in men than women. Risk is increased by family history, and AUD is more common in certain ancestry groups as described in clinical epidemiology summaries.
Patients may present with a wide range of physical and clinical findings related to intoxication, chronic use, or complications. Common clinical clues include recurring minor trauma, gastrointestinal bleeding from gastritis or varices, pancreatitis (acute or chronic), liver disease, alcohol odor on breath, tremulousness, tachycardia, peripheral neuropathy, and recent memory loss. Etiology is multifactorial, with both genetic and social determinants contributing. Commonly described risk factors include unstable social environments, unemployment, divorce, recurrent depression, addiction to other substances (including tobacco), and occupational patterns such as working very long hours.
Diagnosis and initial evaluation rely heavily on screening and history. Screening tools commonly used include CAGE, AUDIT-C, TWEAK/TACE (especially in pregnancy), and CRAFFT (adolescents). CAGE asks about the need to cut down, annoyance by criticism, guilt, and eye-openers; a positive screen should prompt further assessment. AUDIT-C focuses on alcohol frequency and quantity and provides a structured score (0–12), with ≥4 in men and ≥3 in women suggesting problematic drinking requiring more evaluation. A single-question screen (“When was the last time you had more than X drinks in a day?” where X is 5 for men and 4 for women) can also identify unhealthy use when anchored to a recent timeframe. Laboratory tests alone do not diagnose AUD but are useful for identifying complications and nutritional deficiencies. Common findings include elevated gamma-glutamyltransferase (GGT), abnormal AST/ALT (which may be normal or low in advanced liver disease), low albumin, electrolyte abnormalities (hypophosphatemia, hypomagnesemia), macrocytosis with elevated MCV on CBC, positive stool occult blood when gastritis/varices are present, and low vitamin levels including folate, B12, B6, and especially thiamine (B1). Imaging is not routinely required unless trauma is suspected; abdominal ultrasound or CT may show fatty liver or cirrhosis in advanced disease.
Nonpharmacologic treatment is a foundation of care and includes structured psychosocial therapies such as twelve-step facilitation, cognitive behavioral therapy, and motivational enhancement therapy. Coexisting depression should be treated alongside alcohol cessation rather than delayed, because mood disorders commonly coexist and influence relapse risk. Detoxification is not sufficient as stand-alone care and should function as a bridge into a longer-term treatment plan.
Alcohol withdrawal syndrome is managed based on severity and timing from the last drink. Blood ethanol typically declines at about 20 mg/dL per hour. Early withdrawal (“tremulous state”) often begins within 6 to 8 hours after the last drink (or 12 to 48 hours after reduction), peaking around 24 to 36 hours, with tremor, insomnia, mild agitation, and tachycardia. Depending on support system, comorbidities, and risk factors for severe withdrawal, detoxification may occur in outpatient or inpatient settings. Inpatient care includes monitoring (often with standardized withdrawal charts), seizure precautions, and symptom-titrated sedation. Benzodiazepines are the cornerstone of withdrawal management and are used either as fixed-dose tapers or symptom-triggered regimens. Symptom-triggered protocols often use the CIWA-Ar scale to guide dosing; patients with mild symptoms can sometimes be monitored outpatient, while higher CIWA-Ar scores and high-risk histories warrant inpatient detoxification. Beta-blockers can help control tachycardia and hypertension but should not be used alone because they do not prevent seizures or delirium; clonidine can reduce autonomic symptoms in mild to moderate cases but similarly does not prevent seizures or delirium. Vitamin replacement is essential, especially thiamine, and thiamine should be administered before IV dextrose to reduce the risk of precipitating Wernicke encephalopathy. Hydration and correction of electrolytes (including magnesium and phosphate) are often required.
Alcoholic hallucinosis consists of predominantly auditory hallucinations (sometimes visual/tactile/olfactory) that often peak 24 to 36 hours after abstinence and can resemble a primary psychotic disorder, but typically occurs without the marked clouding of sensorium seen in delirium tremens. Withdrawal seizures usually occur within 7 to 30 hours after cessation (often peaking around 13 to 24 hours) and are typically generalized; focal deficits, prolonged confusion, or fever should prompt evaluation for alternative causes (including imaging and lumbar puncture when indicated). Delirium tremens (DTs) is the most severe withdrawal state, usually occurring within the first week after reduction or cessation of heavy intake, commonly peaking around 72 to 96 hours, and characterized by profound confusion, tremor, vivid hallucinations, and marked autonomic hyperactivity; untreated mortality is significant. DTs require close monitoring, aggressive supportive care, and adequate benzodiazepine sedation (sometimes with agents such as lorazepam, chlordiazepoxide, diazepam, or midazolam in titratable settings), along with correction of fluids, electrolytes, and coexisting medical problems.
Long-term (chronic) treatment focuses on relapse prevention and sustained recovery using both psychosocial support and, when appropriate, pharmacotherapy. Acamprosate supports maintenance of abstinence after detoxification and is typically dosed as 666 mg three times daily; it should be avoided in severe renal impairment. Naltrexone (oral or extended-release monthly injection) reduces the rewarding effects of alcohol and can reduce heavy drinking; it must be avoided in acute hepatitis/hepatic failure and in people using opioids, as it can precipitate opioid withdrawal. Disulfiram causes an aversive reaction to alcohol ingestion by blocking aldehyde dehydrogenase, but is now used less often. Agents such as topiramate, baclofen, or gabapentin may be considered second line in moderate to severe AUD, with some guidance suggesting topiramate may be preferable in certain cases. Pharmacologic therapy is generally avoided in pregnant or breastfeeding individuals.
Disposition and referral are key components of management. Community supports such as Alcoholics Anonymous, Adult Children of Alcoholics, and family supports like Al-Anon or Al-A-Teen can be helpful. Inpatient detoxification is relatively indicated for patients with a history of DTs or withdrawal seizures, severe withdrawal symptoms, significant psychiatric or medical comorbidities, pregnancy, multiple prior detoxifications, recent high levels of alcohol intake, or lack of a reliable support network. Even when detoxification is successful, long-term outcomes depend heavily on engagement in ongoing treatment and social support, because relapse is common without sustained intervention.

- Published on
KembaraXtra- Medicine – Alcoholic Hepatitis
Alcoholic hepatitis (AH) is a severe, progressive inflammatory and cholestatic liver disease that occurs in the setting of long-term heavy alcohol use—commonly described as roughly 60–80 g/day in men and 20–40 g/day in women. It is characterized by a rapid onset of jaundice, hepatomegaly, generalized malaise, and features of a systemic inflammatory response. Patients often have a long history of heavy intake, frequently exceeding 100 g of alcohol daily for two or more decades, with a typical presentation age in the 40s to 50s, usually before age 60. While AH is more common in men overall, women may develop AH after a shorter duration and lower cumulative exposure. Risk factors described include poor nutrition, obesity, female sex, Hispanic ethnicity, drinking multiple alcohol types, and drinking between meals.
Clinically, AH often presents with jaundice developing within about 60 days of heavy alcohol consumption (often >50 g/day for at least 6 months), with jaundice duration typically less than 3 months. Patients may report right upper quadrant or epigastric pain, nausea or vomiting, malaise, anorexia, low-grade fever, abdominal distention (often from ascites), weight loss or malnourishment, and proximal muscle wasting or weakness. With worsening hepatic dysfunction, complications can include gastrointestinal bleeding and encephalopathy (confusion, lethargy). On examination, common findings include jaundice, ascites, a tender enlarged liver, tachycardia or tachypnea, hypotension, peripheral edema, shifting dullness from ascites, splenomegaly, and sometimes a hepatic bruit. If cirrhosis coexists, stigmata such as gynecomastia, spider angiomata, altered hair distribution, and muscle wasting may be present. Fever should prompt evaluation for infection, including spontaneous bacterial peritonitis, urinary tract infection, and pneumonia.
Diagnosis is primarily clinical and supported by characteristic laboratory patterns, while excluding competing causes of acute hepatitis and cholestasis such as hepatitis B or C, nonalcoholic steatohepatitis, drug-induced liver injury, hemochromatosis, and cholangitis. A detailed alcohol history is essential, and screening for alcohol misuse can be supported with tools such as the AUDIT questionnaire. Laboratory findings commonly include AST elevation (>45 U/L but typically <500 u />) with an AST:ALT ratio ≥2:1, total bilirubin often >5 mg/dL, and prolonged PT/INR. Additional supportive findings may include elevated GGT, elevated CRP, electrolyte and micronutrient abnormalities (such as hypokalemia, hypomagnesemia, low zinc, hypophosphatemia), hypoalbuminemia, hyperferritinemia, and CBC abnormalities (leukocytosis with bandemia, anemia, thrombocytopenia, and an elevated MCV). Testing is also used to rule out alternative diagnoses, including viral hepatitis serologies and other targeted labs (e.g., ferritin/transferrin saturation, alkaline phosphatase, alpha-fetoprotein when appropriate). Imaging is used both to evaluate liver morphology and to exclude other causes; abdominal ultrasound is typically the imaging study of choice to assess for gallstones, biliary obstruction, liver abscess, or hepatocellular carcinoma. Liver biopsy is rarely required, but may be helpful when the diagnosis is uncertain, to assess coexisting disease, or to exclude alternative pathology; typical histology can include steatosis, hepatocyte ballooning and necrosis, Mallory–Denk bodies, perivenular fibrosis, and lobular/portal inflammation.
Management is commonly framed around three pillars: (1) severity assessment, (2) supportive care with abstinence and nutrition, and (3) pharmacologic therapy for severe disease when indicated. Severity is often assessed with scoring systems such as Maddrey Discriminant Function (MDF) and MELD, and sometimes the Glasgow alcoholic hepatitis score. Severe AH is often defined by MDF ≥32, MELD >20–21, and/or presence of hepatic encephalopathy; these thresholds are associated with high short-term mortality and generally warrant hospitalization. Abstinence from alcohol, together with nutritional support, is the cornerstone of treatment and improves both short- and long-term survival. Nonpharmacologic supports for abstinence include cognitive behavioral therapy, motivational interviewing, and Alcoholics Anonymous attendance, while pharmacologic relapse-prevention aids may include agents such as naltrexone, acamprosate, or baclofen when appropriate. Monitoring abstinence can include breath testing, urine drug screening for recent use, or hair testing for ethyl glucuronide for longer windows, depending on clinical context. Smoking cessation and treatment of other substance use are also recommended to reduce oxidative stress and improve outcomes.
Nutrition is emphasized because many patients have protein-calorie malnutrition and vitamin/mineral deficiencies. Support commonly includes liberal vitamin supplementation (notably thiamine, folate, and vitamin K), mineral supplementation (generally avoiding iron unless specifically indicated), careful calorie counting with increased caloric targets, and protein intake around 1.2–1.5 g/kg/day of ideal body weight, with possible temporary restriction only in severe encephalopathy. Fluid management is individualized, especially in patients with ascites or renal dysfunction.
For severe alcoholic hepatitis, glucocorticoids may be used in selected patients—commonly prednisolone 40 mg/day for 28 days followed by a taper—aiming to reduce hepatic inflammation and injury. Steroid therapy is not universally beneficial in all studies and must be weighed against contraindications such as active infection or gastrointestinal bleeding. Clinical response should be reassessed during treatment, and therapy is typically discontinued in nonresponders based on bilirubin trends and prognostic models. For patients who cannot receive corticosteroids, pentoxifylline has been used historically, although evidence is limited and it is not effective as rescue therapy for steroid nonresponders.
Liver transplantation can be considered in carefully selected patients with severe disease (e.g., very high MELD and poor response to medical therapy). Early liver transplantation has been associated with favorable survival in selected candidates, though programs apply strict psychosocial evaluation and relapse-risk assessment. Historically, many centers required 6 months of sobriety, but selection is increasingly individualized and not based solely on time abstinent. Severe AH may require ICU-level care and multidisciplinary referral, including gastroenterology/hepatology, nutrition services, nephrology for hepatorenal syndrome or renal failure, infectious disease for fever/leukocytosis, and neurology for altered mental status or seizures. Ongoing follow-up includes monitoring metabolic panels and liver tests, reinforcing abstinence, and—if cirrhosis is present—surveillance strategies such as periodic alpha-fetoprotein testing and liver ultrasound for hepatocellular carcinoma, along with vaccination against hepatitis A and B and routine adult immunizations when appropriate.

- Published on
KembaraXtra-Medicine – Anaerobic Infections
Anaerobic infection is caused by bacteria that require a reduced oxygen tension for growth. Relevant ICD-10CM codes include A41.4 (Sepsis, anaerobic) and A48 (Cellulitis, anaerobic).
Anaerobic infections may occur at any site, but most are anatomically related to mucosal surfaces. They should be suspected when there is foul-smelling tissue, soft tissue gas, necrotic tissue, or abscess formation. In the head and neck, odontogenic infections from dental or soft tissue sources may progress to periapical abscesses and sometimes extend to bone. Chronic sinusitis, chronic mastoiditis, peritonsillar abscess, and chronic otitis media can involve both anaerobic and aerobic pathogens. Complications include deep neck space infections, brain abscesses, and mediastinitis. Specific head and neck examples include Ludwig angina, a bilateral infection of the sublingual and submandibular spaces causing swelling at the base of the tongue with potential airway compromise and usually involving mixed aerobic/anaerobic flora, and Lemierre syndrome, a jugular vein suppurative thrombophlebitis caused by anaerobic bacteria, classically Fusobacterium necrophorum.
In pleuropulmonary disease, infection may involve anaerobes that normally reside in the oropharynx. Aspiration is more common in people with altered mental status or seizures, and anaerobic infection is more likely in those with gingivitis or periodontitis. Manifestations include necrotizing pneumonia, empyema, and lung abscess. In intraabdominal infections, disruption of intestinal integrity can lead to anaerobic infection; bacteria may originate from colonic neoplasm, perforated appendicitis, diverticulitis, or bowel surgery, resulting in bacteremia, peritonitis, and sometimes intraabdominal abscesses. These infections are usually mixed, containing both anaerobes and aerobes. In the female genital tract, anaerobes are involved in bacterial vaginosis, salpingitis, endometritis, pelvic abscesses, and septic abortion, and these infections also tend to be mixed. Pelvic thrombophlebitis may occur when a resolving pelvic infection is accompanied by new or persistent fever.
Other anaerobic infections include skin and soft tissue infections at any site, with more commonly associated entities such as synergistic gangrene, bite wound infections, and infected decubitus ulcers. The clinical significance of anaerobes in diabetic foot infections is unclear. Anaerobic bacteremia is not common; when it occurs the source is most often intraabdominal, followed by female genital tract, pleuropulmonary, and head/neck sources. Anaerobes can also be involved in osteomyelitis, especially when associated with decubitus ulcers or vascular insufficiency, and facial bone osteomyelitis can arise from adjacent infections of teeth or sinuses.
Etiologically, anaerobic infections are most commonly endogenous, arising from bacteria that normally line mucosal surfaces. Infection results when mucosal barriers are disrupted by trauma, ischemia, surgery, or perforation, allowing organisms access to normally sterile sites, causing tissue destruction and abscess formation. Synergy between different anaerobes, or between anaerobes and aerobes, is important. Examples of anaerobic bacteria include gram-negative organisms such as Bacteroides spp., Fusobacterium, and Prevotella spp., and gram-positive organisms such as Peptostreptococcus, Clostridioides spp., Finegoldia magna, and Actinomyces spp.
The main differential diagnosis is an aerobic bacterial infection without anaerobes. Another key differential is ischemic necrosis without infection, distinguishing “dry” gangrene (noninfected necrosis) from “wet” gangrene (infected tissue with anaerobic infection).
Workup requires attention to specimen handling: samples submitted for anaerobic culture should be processed within 30 minutes, and growth may take 5–7 days. A large volume of material is more likely to yield significant growth; swabs are less efficient for transporting infected material. Blood cultures should be obtained, preferably before antibiotics are given. Laboratory findings may include elevated WBC count (extremely high counts can be seen with pseudomembranous colitis), positive stool C. difficile testing by PCR/NAAT, increased lactate levels in ischemia or perforation, and possible positive blood or wound cultures—though failure to grow anaerobes is common due to inadequate culturing techniques or fastidious organisms. Imaging may include plain films to show gas in tissues, free air from perforated viscus, or air/fluid levels within an abscess, and ultrasound/CT/MRI to identify abscesses or tissue destruction.
Treatment includes nonpharmacologic measures such as removal of necrotic tissue and drainage of abscesses, often accomplished via CT-guided percutaneous drainage. Oral antibiotic options with anaerobic activity include clindamycin, metronidazole, and chloramphenicol, with broader coverage from amoxicillin/clavulanate; penicillin VK is used for odontogenic infections. For C. difficile–associated diarrhea, oral vancomycin is now the preferred agent, while metronidazole is considered second-line; oral vancomycin is used for subsequent episodes and for pulse or taper regimens. For more serious illness, parenteral options include IV clindamycin, metronidazole, and chloramphenicol; cephalosporins with anaerobic or mixed coverage such as cefoxitin and cefotetan; extended-spectrum penicillins (e.g., piperacillin) and beta-lactam/beta-lactamase inhibitor combinations with significant anaerobic activity and varying broad-spectrum coverage, including ampicillin/sulbactam, ticarcillin/clavulanate, and piperacillin/tazobactam; and carbapenems such as imipenem, meropenem, doripenem, or ertapenem, which have extensive anaerobic activity. Actinomycosis is treated with penicillin for 6–12 months. Sulfamethoxazole-trimethoprim and fluoroquinolones are generally ineffective, though some newer quinolones such as moxifloxacin have inhibitory activity against anaerobes.
Disposition emphasizes that all necrotic debris must be removed or the infection will recur, and follow-up is critically important to ensure resolution. Referral to a surgeon is appropriate if drainage is required, and infectious disease consultation may be helpful in complicated patients or when the regimen is failing or response is slow. Resistance rates are generally low for many agents used against anaerobes (reported rates: piperacillin/tazobactam 0.5%, ampicillin/sulbactam 3%), though one study found >50% resistance of Bacteroides fragilis to clindamycin.
- Published on
KembaraXtra-Medicine – Amphetamine Use Disorder
Amphetamine use disorder is a pattern of amphetamine use marked by impairment in control of use or social obligations, risky use, or physical dependence within a 12-month period. Severity is categorized as mild, moderate, or severe based on the number of symptoms. Amphetamines are central nervous system stimulants that increase monoamine neurotransmitters (dopamine, norepinephrine, serotonin) and are defined by a phenylethylamine structure. Amphetamine and dextroamphetamine are FDA approved for AD/HD, narcolepsy, and obesity. Methamphetamine is a type of amphetamine that is FDA approved for AD/HD and obesity, but is more commonly illicitly produced and used recreationally. Synonyms include stimulant use disorder and methamphetamine use disorder. ICD-10CM codes include F15.10 (mild), F15.20 (moderate or severe), F15.11 (mild, in early or sustained remission), and F15.21 (moderate or severe, in early or sustained remission).
Epidemiologically, prevalence is described as rare: 0.6% among people ages ≥12 years (about 1.5 million) with methamphetamine use disorder, and 0.3% among people ≥12 years (about 758,000) with prescription stimulant use disorder. Among adults who used methamphetamine in the past year, 53% met criteria for methamphetamine use disorder. Methamphetamine use disorder is more common among those age 26 and older (0.6%) than under age 25, while prescription stimulant use disorder is more common between ages 12 and 25 compared with ≥26.
Risk increases in people with any substance use disorder. Among psychiatric comorbidities, bipolar disorder, schizophrenia, and antisocial personality disorder are associated with stimulant use disorder. Demographic risk factors for methamphetamine use include male sex, lower educational attainment, and non-Hispanic White ethnicity. Methamphetamine use is higher among men who have sex with men, including in “chemsex,” where methamphetamine and other stimulants are used to increase arousal and reduce inhibition during sexual sessions.
Clinically, amphetamine use disorder can develop as quickly as within 1 week of exposure, and most people experience physical dependence in the form of tolerance or withdrawal. Amphetamines may be used by oral ingestion, insufflation, injection, or smoking, and use is often described as chronic or episodic binges. During intoxication, patients may report euphoria and may present with hyperarousal, aggressive behavior, psychosis (paranoia and hallucinations), panic attacks, or suicidal and homicidal ideation. With toxicity, sympathomimetic signs such as hypertension, tachycardia, dilated pupils, and flushed skin can occur. Some intoxicated patients report chest pain, and amphetamine use increases risk of myocardial infarction, arrhythmias, and strokes. During withdrawal, patients commonly report depressive symptoms. Risk for psychosis increases with higher frequency and amount of use, and psychosis can occur within months to several years after starting amphetamine use.
The exact cause of amphetamine use disorder is not known, but amphetamine-type medications act on the dopamine and monoamine transporter (DAT and VMAT2), increasing dopamine transmission in the mesolimbic dopamine pathway (the reward pathway).
In diagnosis, amphetamine use disorder is different from amphetamine misuse, which refers to use inconsistent with prescription guidelines. Patients using PCP or novel psychoactive substances (such as cathinones/bath salts) may present similarly and can be distinguished with a toxicology test. Primary psychiatric disorders (e.g., schizophrenia) or depressive disorders (e.g., major depressive disorder) can be evaluated by establishing a timeline of symptoms relative to amphetamine use; psychosis that quickly improves is more consistent with amphetamine-induced psychosis.
Work-up may be difficult because detailed history can be hard to obtain during acute intoxication or withdrawal. An ECG should be performed in patients with tachycardia. A safety assessment is essential to determine whether the patient can safely care for themselves, especially when suicidal or homicidal ideation is reported. Recommended labs include basic testing such as Chem 7, serum lactate, creatine phosphokinase, thyroid function tests, and hepatic enzymes. A urine or serum toxicology test can confirm amphetamine presence. There are no radiographic tests for diagnosis of stimulant use disorder, but CT imaging may be considered for strokes or vasculopathies associated with amphetamine use.
Treatment is primarily nonpharmacologic, with behavioral interventions as the mainstay. Approaches such as contingency management, cognitive-behavioral therapy, behavioral activation, and exercise have shown mild effectiveness in reducing symptoms and sustaining abstinence. Contingency management, especially when combined with community reinforcement (reorganizing the environment so abstinence is more rewarding than drug use), has been particularly shown to be effective.
For acute general management, antipsychotics—especially olanzapine and haloperidol—have evidence for treating amphetamine-associated psychosis. No medication has been shown to be effective for amphetamine withdrawal. There is limited evidence that amineptine (a tricyclic antidepressant no longer available due to abuse potential) reduced discontinuation symptoms and improved clinical presentation, and there is mixed evidence that mirtazapine may reduce anxiety symptoms during withdrawal.
For chronic management, there is no evidence that pharmacologic treatment alone is sufficient. Randomized controlled trials show limited benefit from methylphenidate, bupropion, modafinil, and naltrexone in reducing amphetamine use.
Disposition-wise, methamphetamine use disorder often follows a course of intense use, sobriety, and relapse. Complications of amphetamine intoxication (including ecstasy) include hyponatremia due to SIADH (very rare; managed with water restriction), intracerebral hemorrhage (uncommon; CT/MRI if focal signs or reduced GCS), serotonin toxicity (uncommon; supportive care and cyproheptadine for serious cases), hypertension (uncommon; GTN infusion, benzodiazepines, labetalol; avoid beta-blockers), hyperthermia (common; cooling measures, diazepam, cyproheptadine), rhabdomyolysis (uncommon; IV fluids and cooling), supraventricular and ventricular tachycardia (uncommon; short-acting antiarrhythmics such as esmolol for SVT), and seizures (uncommon; IV diazepam).
Referral is typically to physicians and mental health professionals. Important pearls include that stimulant use disorders often co-occur with sedative substance use disorders (e.g., alcohol, marijuana, opioids) because these may relieve stimulant side effects such as insomnia and anxiety. Amphetamine use is also associated with an increased risk of death by suicide, so assessing suicidality is critical in any patient presenting with amphetamine use.
For prevention, community-based universal prevention programs that promote youth social-emotional skills and reduce risk factors have been shown to lead to sustained reductions in lifetime methamphetamine use.

- Published on
KembaraXtra-Medicine – Amyloidosis
Amyloid is an altered, insoluble protein folded in β-pleated sheets that is deposited extracellularly. Amyloid can accumulate in one or many organs, causing organ dysfunction. Amyloidosis refers to a heterogeneous group of disorders characterized by deposition of an amorphous, extracellular fibrillar protein in various organs and tissues. Subtypes include primary amyloidosis (AL), secondary amyloidosis (AA), hereditary amyloidosis, and localized amyloidosis. ICD-10CM codes include E85.0 (non-neuropathic heredofamilial), E85.1 (neuropathic heredofamilial), E85.2 (heredofamilial, unspecified), E85.3 (secondary systemic), E85.4 (organ-limited), E85.8 (other), and E85.9 (unspecified).
Amyloidosis nomenclature is based on the amyloid protein precursor and associated clinical features. AL or AH derives from immunoglobulin light or heavy chain and is primary or localized, often associated with myeloma or macroglobulinemia. AA derives from serum amyloid A (SAA) and occurs as secondary amyloidosis or with familial Mediterranean fever and other familial periodic fever syndromes. ATTR derives from transthyretin and is familial or senile. A fibrinogen derives from fibrinogen and is associated with familial renal amyloidosis (Ostertag type). Aβ2M derives from β2-microglobulin and is dialysis-associated, including carpal tunnel syndrome. Aβ derives from amyloid-β precursor protein (ABPP) and is associated with Alzheimer disease. Apo A-I/A-II–related forms include apolipoprotein A-I (proteinuria) and apolipoprotein A-II (cardiac and neuropathy patterns). A lysozyme involves lysozyme deposition affecting the GI tract, liver, and kidneys. ALECT2 is noted as a renal form (leukocyte chemotactic factor 2 amyloidosis).
A major classification system (not exhaustive) includes: AA (serum amyloid A) causing systemic amyloidosis, often predominantly renal, linked to acquired or hereditary chronic inflammatory disease (formerly “secondary/reactive”); AL (monoclonal immunoglobulin light chains) causing systemic amyloidosis potentially involving many organs and associated with myeloma/monoclonal gammopathy/occult B-cell dyscrasias (formerly “primary”); and ATTR wild-type (normal plasma transthyretin) causing nonhereditary systemic amyloidosis with predominantly cardiac involvement (formerly “senile cardiac amyloidosis”). ATTR variant (genetic variants such as ATTR Met30, Ala60, Ile122) can cause familial amyloid polyneuropathy (FAP) often with prominent cardiomyopathy, and certain mutations (e.g., TTR Ile122) may produce predominantly cardiac involvement without neuropathy. Aβ2M (β2-microglobulin) causes dialysis-related amyloidosis with renal failure and long-term dialysis, with predominantly articular/periarticular involvement. Aβ causes cerebrovascular and intracerebral plaque amyloid in Alzheimer disease and may be familial. AApoAI (genetic variants of apolipoprotein AI) causes autosomal dominant systemic amyloidosis, typically nonneuropathic with visceral involvement—especially nephropathy—and minor wild-type deposits can occur in the aorta with aging. AApoAII causes autosomal dominant systemic amyloidosis with predominantly renal involvement. AFib (fibrinogen A α-chain variants) causes autosomal dominant systemic amyloidosis with nonneuropathic predominant nephropathy. ALys (lysozyme variants) causes autosomal dominant systemic amyloidosis with predominantly renal and GI involvement and can rarely present with hepatic rupture. ACys (cystatin C variant) is linked to hereditary cerebral hemorrhage with cerebral and systemic amyloidosis in Icelandic individuals. AGel (gelsolin variants) causes autosomal dominant systemic amyloidosis with predominantly cranial nerve involvement plus lattice corneal dystrophy and is described as most common in Finland.
In the United States, incidence is estimated at 1500–3500 new cases annually, with AL being the most common type. Amyloidosis primarily affects men ages 60–70 years. Common presenting symptoms include fatigue, dyspnea, edema, paresthesias, and weight loss, and other findings depend on organ involvement. Diagnosis should be considered in patients with a multisystem disorder involving the heart, kidney, liver, or nervous system. Renal involvement may present with nephrotic syndrome. Pulmonary involvement may cause fatigue and dyspnea. GI involvement is uncommon but may present with diarrhea, nausea, abdominal pain, and macroglossia. Cardiac involvement produces an infiltrative cardiomyopathy with preserved ejection fraction and diastolic dysfunction; in transthyretin-related disease, transthyretin dissociation leads to deposition of misfolded monomers as amyloid fibrils in the myocardium with subsequent cardiomyopathy and heart failure. Patients may have bleeding problems from factor X deficiency or fragile vessels infiltrated by amyloid; periorbital bleeding (“raccoon eyes”) is characteristic. Nervous system involvement can cause peripheral neuropathy, tendinopathy, muscle weakness, numbness, syncope, or dizziness, and associated autonomic neuropathy can be severely disabling. Syndromes in primary (AL) amyloidosis include nephrotic/nephrotic with renal failure (30%), hepatomegaly (24%), congestive heart failure (22%), carpal tunnel (21%), neuropathy (17%), and orthostatic hypotension (12%).
The common mechanism is deposition of Congo red–staining amyloid fibrils, but key subtype differences exist. AL is associated with an underlying clonal plasma cell disorder producing abnormal light chains with possible multi-organ deposition. AA has no plasma cell disorder and results from long-standing systemic inflammation (examples listed include tuberculosis, leprosy, malaria, untreated syphilis). Localized amyloidosis results from localized fibril synthesis without a plasma cell disorder. Familial amyloidosis includes forms due to transthyretin gene (TTR) mutations, with hepatocyte-derived transthyretin deposition in peripheral nerves and the heart.
Differential diagnosis depends on organ involvement. For renal disease consider toxin- or drug-induced necrosis, glomerulonephritis, renal vein thrombosis. For pulmonary disease consider sarcoidosis, connective tissue disease, infectious causes. For restrictive cardiomyopathy consider endomyocardial fibrosis or viral myocarditis. For carpal tunnel consider rheumatoid arthritis, hypothyroidism, overuse. For peripheral neuropathy consider alcohol abuse, vitamin deficiencies, diabetes mellitus.
Workup includes blood and urine testing for abnormal light chains, tests for target-organ damage, and histologic confirmation. Typical confirmation is via fat pad aspiration and bone marrow biopsy with Congo red staining. Recommended diagnostic evaluation includes CBC; chemistries and other labs (sodium, potassium, alkaline phosphatase, calcium, phosphorus, AST, bilirubin, creatinine, β2-microglobulin, glucose, cholesterol, uric acid, thyroid profile); serum and urine immunofixation; assays for immunoglobulin free light chains and immunoglobulins G, A, and M; cardiac markers (troponin, NT-proBNP); coagulation evaluation (factor X and prothrombin time); and cardiac/pulmonary assessment with chest x-ray, ECG, and echocardiogram with Doppler and strain imaging. Amyloid staging assigns 1 point each for FLC-diff ≥18 mg/dL, cTnT ≥0.025 ng/mL, and NT-proBNP ≥1800 pg/mL, producing stages I–IV (scores 0–3). Reported median survival (months) by stage is 94.1, 40.3, 14, and 5.8, respectively. Laboratory testing also includes SPEP/UPEP with immunofixation for light chains, routine labs such as BUN/creatinine, liver function tests, thyroid function, urine albumin, and biopsy confirmation; if fat pad biopsy is negative, biopsy of the affected organ may be required.
Imaging can help define organ involvement. Cardiac MRI may show a distinctive pattern. Two-dimensional Doppler echocardiography is useful for cardiac evaluation but is less sensitive than cardiac MRI. Nuclear imaging with technetium-labeled aprotinin may detect cardiac amyloidosis. Labeled diphosphonates are important for typing amyloidosis and diagnosing heart involvement in transthyretin cardiac amyloidosis, and bone scintigraphy may detect cardiac involvement earlier than echocardiography in transthyretin patients. Serum amyloid P (SAP) scintigraphy has high sensitivity for detecting deposits in liver, spleen, kidneys, adrenal glands, bones.
Acute/general management and disease-directed therapy depend on subtype and organ involvement. Patients with AL amyloidosis should be evaluated for autologous hematopoietic stem cell transplantation (HSCT); good candidates include those with good performance status, age <70, and limited organ involvement. tafamidis is an oral transthyretin stabilizer fda approved for adults with transthyretin-mediated amyloid cardiomyopathy (attr-cm) has been shown to decrease hospitalizations deaths in attr-cm. al, chemotherapy aims reduce production of amyloidogenic light chains by targeting clonal plasma cells; agents used multiple myeloma are effective, including melphalan prednisone, imids (thalidomide, lenalidomide) proteasome inhibitors also used. patients ineligible autologous hsct should receive melphalan- or bortezomib-based regimens. trials oligonucleotide drugs (inotersen, patisiran) have improvement clinical manifestations hereditary amyloidosis. who develop renal failure may require hemodialysis transplant. liver transplantation successfully familial secondary (aa) amyloidosis, recognition treatment the underlying disorder needed. newly diagnosed al show that adding daratumumab bortezomib, cyclophosphamide, dexamethasone associated higher hematologic complete response rates.< />pan>
Major treatment options include, for AL amyloidosis, IV melphalan with autologous stem cell rescue (G-CSF mobilized peripheral blood stem cell collection, IV melphalan 140–200 mg/m², autologous stem cell reinfusion), cyclic oral melphalan and dexamethasone (melphalan 0.22 mg/kg/day ×4 days; dexamethasone 20–40 mg/day ×4 days or weekly; repeat every 4 weeks), immunomodulators (lenalidomide 5–15 mg/day ×21 days with dexamethasone 20–40 mg weekly; repeat every 4 weeks), and proteasome inhibitors (IV bortezomib 0.7–1.6 mg/m² 1–2 times per week; repeat every 3–5 weeks). For AA amyloidosis, options include aggressive treatment of underlying inflammatory disease, medical or surgical treatment of infection, colchicine 1.2–1.8 mg/day for AA secondary to familial Mediterranean fever, and investigational antifibril drug eprodisate. For ATTR, options include orthotopic liver transplantation, transthyretin stabilizers (tafamidis, investigational diflunisal), and investigational oligonucleotide drugs (inotersen, patisiran).
Supportive treatment across all amyloidosis types is symptom- and organ-directed. For cardiac congestive failure, options include salt restriction 1–2 g/day and diuretics (furosemide, spironolactone, metolazone). For arrhythmias, consider pacemaker, automatic implantable cardiac defibrillator, and antiarrhythmics. For renal nephrotic syndrome, use salt restriction 1–2 g/day, elastic stockings and leg elevation, maintaining dietary protein, and an ACE inhibitor if blood pressure tolerates. For renal failure, use dialysis (long-term ambulatory peritoneal dialysis or hemodialysis). For autonomic nervous system orthostatic hypotension, options include midodrine, increased dietary salt or fludrocortisone depending on edema, and elastic stockings. For gastric atony/ileus, use small frequent low-fat feedings (6/day), oral nutritional supplements, jejunostomy tube feeding, or parenteral nutrition. For GI diarrhea, options include a low-fat diet (≤40 g), psyllium, loperamide, tincture of opium, and parenteral nutrition. For macroglossia, use a soft solid diet; partial glossectomy is rarely effective. For peripheral sensory neuropathy, avoid trauma and use medications such as gabapentin 100–300 mg three times daily, amitriptyline 25–50 mg at bedtime, or pregabalin 50–100 mg three times daily. For motor neuropathy with footdrop, use ankle-foot orthotics and physical therapy. For hematologic issues with intracutaneous bleeding, avoid trauma and antiplatelet agents; for factor X deficiency, use factor replacement (recombinant factor VIIa, prothrombin complex concentrates). Splenectomy may be considered for splenomegaly.
Prognosis is determined primarily by cardiac involvement and the amyloidosis form. In endomyocardial biopsy–documented cardiac amyloidosis, longer-term survival correlates more strongly with NYHA functional class than with ECG or echocardiographic variables. In AA amyloidosis, eradication of the predisposing disease slows and can occasionally reverse progression, with a median survival after diagnosis of 133 months. Patients with familial amyloidotic polyneuropathy often have a prolonged course of 10–15 years. Dialysis-associated amyloidosis progression can be improved with newer dialysis membranes that pass β2-microglobulin. Median survival in patients with overt congestive heart failure is approximately 6 months, compared with 30 months without CHF.

- Published on
KembaraXtra-Medicine – Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis (ALS) is a progressive and fatal degenerative neuromuscular condition of undetermined etiology that affects the corticospinal tracts and anterior horn cells, resulting in dysfunction of both upper motor neurons (UMNs) and lower motor neurons (LMNs). Synonyms include Lou Gehrig disease, ALS, and motor neuron disease. The ICD-10CM code is G12.21 (Amyotrophic lateral sclerosis).
ALS incidence is about two new cases per 100,000 persons per year. Onset is usually between 50 and 70 years, and the male-to-female ratio is 1.8:1. The estimated prevalence is 5.2 per 100,000 population.
Clinical presentation typically includes a combination of LMN signs—weakness, hypotonia, muscle wasting, fasciculations, and hyporeflexia or areflexia—and UMN signs—loss of fine motor dexterity, spasticity, extensor plantar responses, hyperreflexia, and clonus. Extraocular movements, sensation, and bowel and bladder function are typically preserved. Patients may develop dysarthria, dysphagia, pseudobulbar affect, and frontal lobe dysfunction. Respiratory insufficiency progresses to respiratory failure and death, typically late in the disease. Concomitant frontotemporal dementia occurs in about 15%, and executive dysfunction occurs in another 50%. Pseudobulbar affect (inappropriate crying or laughing not triggered by typical emotional cues) is common and treatable.
ALS commonly manifests in one of three onset patterns. Limb onset accounts for 75%–80% of cases and presents with painless muscle weakness, clumsiness (dropping things, stumbling, falling), impaired fine motor skills (e.g., writing), limb stiffness, gait disturbances, and wristdrop or footdrop. Bulbar onset accounts for 20%–25% and presents with dysarthria or voice change, pseudobulbar affect, dysphagia and/or weight loss, drooling, and choking during swallowing. Respiratory onset occurs in <3% and presents with dyspnea on exertion or at rest, orthopnea, daytime sleepiness, difficulty sleeping, other symptoms of respiratory compromise. als makes up about 90% adult-onset motor neuron diseases; disease presentations include progressive muscular atrophy, primary lateral sclerosis, bulbar palsy, pseudobulbar als-parkinsonism-dementia complex.< />pan>
Regarding etiology and classification, 90% of cases are sporadic and 10% are familial. Known genetic mutations include C9orf72 hexanucleotide repeat expansion (responsible for ~40% of familial ALS among those of European origin and nearly 10% of sporadic ALS, and virtually all ALS cases associated with frontotemporal dementia), SOD1 (copper-zinc superoxide dismutase) mutations (~12% of familial cases), TAR DNA-binding protein mutations (~4% of familial cases), and FUS (~4% of familial cases, rarely associated with frontotemporal dementia), among many others. Risk is increased in Whites, non-Hispanics, age >60 years, family history of ALS, and prior exposure to heavy metals, pesticides, or BMAA. A practical classification includes sporadic/acquired ALS variants (classic spinal-onset, Mills hemiplegic variant, pseudoneuritic, flail-arm, monomelic, UMN onset, LMN onset, bulbar onset, dyspnea onset, progressive muscular atrophy, primary lateral sclerosis, progressive bulbar palsy, Western Pacific ALS) and multiple familial ALS subtypes (ALS1 through ALS25 and additional rare-mutation forms). An FTD-ALS overlap is also recognized (e.g., ALS-FTD associated with C9orf72; and FTD with some ALS features associated with tau and progranulin genes on chromosome 17).
The differential diagnosis includes multifocal motor neuropathy with conduction block; cervical spondylotic myelopathy with polyradiculopathy; spinal stenosis with lumbosacral nerve root compression; chronic inflammatory demyelinating polyneuropathy with CNS lesions; syringomyelia; syringobulbia; foramen magnum tumor; meningeal carcinomatosis; spinal muscular atrophy; polyglucosan body disease; bulbospinal muscular atrophy (Kennedy disease); monomelic amyotrophy; Lyme disease; and ALS-like syndromes reported with lead intoxication, HIV, hyperparathyroidism, hyperthyroidism, lymphoma, and vitamin B12 deficiency.
Diagnosis is based on clinical findings, electromyography results, and exclusion of alternative causes. Workup includes MRI of brain and spine to exclude mimics; electromyography and nerve conduction studies following Revised El Escorial criteria; assessment of respiratory function (forced vital capacity [FVC], negative inspiratory force); assessment of swallowing and nutritional status; and neuropsychologic testing if executive dysfunction or FTD is a concern. The Revised El Escorial criteria require evidence of LMN degeneration (clinical, electrophysiologic, or neuropathologic), evidence of UMN degeneration (clinical), and progressive spread of symptoms/signs within or between regions, together with absence of electrophysiologic/pathologic evidence of another disease explaining UMN/LMN degeneration and absence of neuroimaging evidence of another explanatory disease process. Levels of certainty include Definite ALS (UMN and LMN signs in three regions), Probable ALS (UMN and LMN signs in two regions with UMN signs rostral to LMN signs), Probable ALS—Laboratory Supported (UMN signs in one or more regions with LMN signs by EMG in at least two regions), and Possible ALS (UMN and LMN signs in one region together, or UMN signs in two or more regions, or UMN and LMN signs in two regions without UMN signs rostral to LMN signs).
Laboratory tests that may be considered include vitamin B12, thyroid function, parathyroid hormone, and HIV, plus serum protein electrophoresis with immunofixation. In pure LMN syndromes, consider DNA studies for spinal muscular atrophy, bulbospinal atrophy (Kennedy disease), and hexosaminidase levels (late-onset Tay-Sachs). A 24-hour urine heavy metal screen may be done if indicated. In familial ALS, genetic studies should be performed with appropriate genetic counseling. Imaging is guided by clinical scenario; MRI brain and spinal cord help exclude structural mimics, and a modified barium swallow evaluates aspiration risk.
Treatment includes symptomatic management plus disease-modifying therapy that slows progression in some patients. Symptomatic treatment options span multiple domains: fatigue (pyridostigmine bromide, antidepressants, methylphenidate, amantadine, modafinil; energy conservation, work modification, sleep study with BiPAP if abnormal), spasticity (baclofen, tizanidine, dantrolene sodium, diazepam; physical therapy, range-of-motion exercises, botulinum toxin), jaw clenching (benzodiazepines, botulinum toxin into masseters), cramps (quinine sulfate, baclofen, vitamin E, clonazepam; massage, physical therapy), fasciculations (carbamazepine and reassurance), sialorrhea (hyoscyamine sulfate, diphenhydramine, scopolamine patch, glycopyrrolate, atropine, TCAs; suction machine, botulinum toxin into salivary glands, parotid radiation therapy, steam inhalation, nebulization, dark grape juice), pseudobulbar laughing/crying (TCAs, SSRIs, L-dopa/carbidopa, lithium, mirtazapine, venlafaxine, quinidine/dextromethorphan), thick phlegm (guaifenesin, nebulized N-acetylcysteine, nebulized saline, propranolol; insufflation-exsufflation, high-flow chest wall oscillation therapy, cool mist humidifier, rehydration, pineapple/papaya juice, reduced dairy/caffeine/alcohol), aspiration (cisapride and modified food consistency; in severe cases tracheostomy or modified laryngectomy with tracheal diversion), joint pains (anti-inflammatory drugs, analgesics, range-of-motion and heat), depression (TCAs; SSRIs/venlafaxine/mirtazapine/bupropion; counseling, support groups, psychiatry), insomnia (zolpidem tartrate, lorazepam, opioids, TCAs; pressure air pad/gel mattress; noninvasive ventilation where appropriate), laryngospasm (sublingual lorazepam), respiratory failure (bronchodilators, morphine sulfate; hospital bed, nocturnal noninvasive ventilator, IPPB), and constipation (increase oral fluids, Metamucil, Dulcolax suppositories, lactulose/other laxatives, exercise, “power pudding” of prune juice/prunes/applesauce/bran).
Nonpharmacologic therapy is central. Noninvasive ventilation can improve quality of life and may increase tracheostomy-free survival in patients with respiratory difficulty (orthopnea or FVC ≤50% predicted). Percutaneous endoscopic gastrostomy (PEG) can improve nutrition, stabilize weight, and ease medication administration; some studies suggest it may prolong life by 1–4 months, especially when placed before FVC falls to ≤50% predicted. Supportive services include nutrition support, speech therapy, physical and occupational therapy, suction devices for sialorrhea, cough assist devices for ineffective cough and airway clearance, and computerized assistive communication devices. Early advance-care planning discussions should address living will, resuscitation orders, preferences regarding PEG and tracheostomy, and long-term care options, and patients should be encouraged to connect with local support groups.
Disease-modifying medications do not cure ALS but can slow progression in selected patients. Riluzole (glutamate antagonist) was the first FDA-approved medication shown to extend tracheostomy-free survival; typical dosing is 50 mg every 12 hours, taken at least 1 hour before or 2 hours after meals, and it prolongs survival by about 2–3 months; ALT monitoring is recommended monthly for the first 3 months, then every 3 months until completion of the first year, and periodically thereafter. Edaravone (FDA approved 2017) slowed physical decline by 33% versus placebo in a 6-month phase III trial; it is given IV with an initial cycle of daily infusions for 14 days followed by 14 days off, then subsequent cycles of 10 infusions over 14 days followed by 14 days off; cost and insurance coverage can be limiting. Relyvrio (sodium phenylbutyrate/taurursodiol) is an FDA-approved oral fixed-dose combination (approved 2022) for adults with ALS that helps slow progression; its exact mechanism is unknown, but it targets endoplasmic reticulum and mitochondria-dependent neuronal degeneration pathways, and is thought to reduce neuronal death and dysfunction.
Chronic management includes targeted symptom control such as treating sialorrhea with glycopyrrolate or amitriptyline (and considering propranolol or metoprolol if secretions are thick), using botulinum toxin if refractory; treating spasticity with baclofen, tizanidine, or clonazepam; and treating pseudobulbar affect with amitriptyline, sertraline, or dextromethorphan/quinine.
Disposition reflects the progressive nature of ALS: there is currently no cure, mean symptom duration is 3-5 years, and about 20% of patients survive more than 5 years. Referral to a neurologist experienced in neuromuscular disease is recommended to confirm diagnosis, and multidisciplinary ALS clinics may improve survival. Gastroenterology referral for PEG placement is recommended while FVC remains >50% to reduce procedural morbidity, and pulmonology referral is appropriate for noninvasive ventilation such as BiPAP.
Pearls include the importance of patient-physician communication at diagnosis and throughout care, and the value of a multidisciplinary supportive approach to improve daily functioning and independence. Related content includes patient information resources on ALS.

- Published on
KembaraXtra-Medicine – Anal Fissure
An anal fissure is a tear in the epithelial lining of the anal canal, extending from the dentate line to the anal verge. Acute anal fissures usually heal with conservative treatment within 6 weeks, while chronic fissures typically need a more aggressive approach and may require surgery. Synonyms include anorectal fissure and anal ulcer. The ICD-10CM codes are K60.0 (acute anal fissure), K60.1 (chronic anal fissure), and K60.2 (anal fissure, unspecified).
An estimated 235,000 new cases occur each year in the United States. Anal fissures can happen at any age but are more common in infants and middle-aged adults, and they occur more often in men than women. Women are more likely than men to have an anterior fissure (10% vs 1%, respectively).
Clinically, separating the buttocks typically reveals a tear in the posterior midline or less often the anterior midline. Acute fissures usually cause sharp burning or tearing pain with bowel movements and may produce bright-red blood on toilet paper, as a streak on stool, or in the toilet water; they look like a fresh laceration. Chronic fissures are more associated with perianal pruritus or irritation, less intense pain or no pain, and intermittent bleeding. They may show a sentinel tag at the caudal aspect, a hypertrophied anal papilla proximally, and raised edges that expose the horizontal fibers of the internal anal sphincter, reflecting chronic infection and fibrosis. Atypical fissures (acute or chronic) are more likely to appear off the midline, may extend proximal to the dentate line, may be unusually wide or deep, and may be multiple, recurring, or nonhealing; they can be associated with an edematous, tender perianal tag and are more often linked to an underlying systemic disease.
Most fissures start after passing a large, hard stool, but they can also result from frequent defecation and diarrhea. Infectious causes include bacterial infections such as tuberculosis, syphilis, gonorrhea, chancroid, and lymphogranuloma venereum, and viral infections such as herpes simplex virus, cytomegalovirus, and HIV. Other causes include inflammatory bowel disease (Crohn disease, ulcerative colitis), trauma (e.g., hemorrhoidectomy, foreign bodies, anal intercourse), and malignancy (carcinoma, lymphoma, Kaposi sarcoma).
Important differential diagnoses include proctalgia fugax, thrombosed hemorrhoid, carcinoma, and anal fistula. Evaluation may include a digital rectal examination performed only after fully lubricating the anus with 2% lidocaine jelly and waiting 5–10 minutes, plus anoscopy and proctosigmoidoscopy to exclude inflammatory or neoplastic disease. A biopsy is recommended if there is doubt about the cause, and studies should be done with adequate anesthesia. Colonoscopy is considered if IBD or malignancy is suspected, and a small-bowel series is occasionally obtained for similar reasons.
Nonpharmacologic management includes sitz baths, a high-fiber diet, and increased oral fluids. Acute treatment includes a bulk-forming agent (e.g., Metamucil) and/or a stool softener, and local anesthetic jelly (though it may worsen pruritus ani). Nitroglycerin ointment 0.4% can be used by applying 1 inch of ointment (≈1.5 mg nitroglycerin) intra-anally every 12 hours for up to 3 weeks; this product (Rectiv) can be expensive, and topical diltiazem (pharmacy-compounded) is also effective and usually cheaper. Suppositories are not recommended. If medical therapy fails after 2 months, surgery is considered.
For chronic fissures, medical therapy may still be offered because it is often better tolerated and avoids the risk of fecal incontinence. Options include topical glyceryl trinitrate, topical calcium channel blockers (e.g., topical nifedipine or topical 2% diltiazem cream), and botulinum toxin A injections into each side of the internal anal sphincter; botulinum toxin can heal chronic fissures in more than 90% of patients, although dosing and best injection site are not fully clear and it can be expensive. The standard, durable surgical treatment is lateral internal anal sphincterotomy, which has >90% long-term healing, is generally durable, and is noted not to compromise long-term fecal continence in the provided text—though it can lead to fecal incontinence and therefore requires careful patient selection.
Disposition is typically outpatient surgery when surgery is needed. Referral is recommended if the fissure does not improve with conservative measures in 4–6 weeks, if the patient prefers surgery for an acute fissure, or if the patient has a chronic fissure. A special consideration is that HIV-positive patients should be referred to clinicians experienced with the infectious and neoplastic conditions that can mimic anal ulcers in this population.

- Published on
KembaraXtra-Medicine – Amebiasis
Amebiasis is an infection caused by the protozoal parasite Entamoeba histolytica. Although it primarily involves the colon, the disease may extend beyond the intestine, most notably causing amebic liver abscess, which is the most common and serious extraintestinal manifestation. When the intestinal infection is severe and associated with dysentery, it is also referred to as amebic dysentery. Amebiasis is classified under ICD-10CM codes A06.9 (amebiasis, unspecified), A06.1 (chronic intestinal amebiasis), and A06.7 (cutaneous amebiasis).
From an epidemiologic perspective, the incidence of amebiasis in the United States is approximately 1.2 cases per 100,000 population. Although relatively uncommon, it is clinically significant and occurs most frequently among institutionalized individuals, travelers to or immigrants from developing countries, and individuals engaging in oral–anal sexual practices. The estimated prevalence in the United States is about 4%, with approximately 80% of infections remaining asymptomatic. In contrast, in the developing world, amebiasis continues to be a major cause of morbidity and mortality, accounting for approximately 9% of all deaths in children younger than 5 years of age. The disease affects males and females equally overall; however, there is a marked male predominance in cases of amebic liver abscess. Amebiasis most commonly affects individuals in the second through sixth decades of life, with peak incidence noted in children aged 2 to 3 years and in adults older than 40 years.
The clinical presentation of amebiasis is often nonspecific, and many infected individuals remain asymptomatic. Approximately 20% of patients develop symptoms, most commonly diarrhea, which may be bloody, along with abdominal or back pain. In severe intestinal disease, abdominal tenderness is present in about 83% of cases, and fever occurs in approximately 38%. Patients with amebic liver abscess typically present with hepatomegaly, right upper quadrant abdominal tenderness, and fever, although these findings may be absent in fulminant disease.
The etiology of amebiasis involves infection with E. histolytica, while E. dispar and E. moshkovskii are more common but nonpathogenic species that are morphologically indistinguishable from E. histolytica. Transmission occurs through the fecal–oral route, and infection is highly contagious, requiring ingestion of only a small number of cysts in contaminated food or water. The infection is usually localized to the large intestine, particularly the cecum, where it may form a localized inflammatory mass known as an ameboma. In some cases, the organism invades the intestinal mucosa and gains access to the portal circulation, leading to extraintestinal disease. Important risk factors include travel to endemic regions, residence in institutions for the intellectually disabled, immunodeficiency, and male sex, particularly with respect to liver abscess formation.
The differential diagnosis of severe intestinal amebiasis includes ulcerative colitis and other forms of infectious enterocolitis, such as those caused by Shigella, Salmonella, Campylobacter, or invasive Escherichia coli. In elderly patients, ischemic colitis may present with a similar clinical picture and should also be considered.
The diagnostic workup focuses on stool-based testing and evaluation for extraintestinal disease. Stool antigen testing is more sensitive than routine ova and parasite examination for diagnosing amebiasis. Examination of three stool specimens collected over 7 to 10 days yields a sensitivity of 85% to 95%, although microscopy cannot reliably distinguish pathogenic from nonpathogenic species. Diagnostic yield can be improved by concentrating and staining specimens with Lugol iodine or methylene blue, and fecal leukocytes may or may not be present.
Laboratory testing includes fecal enzyme-linked immunosorbent assay (ELISA) antigen detection, which is specific for E. histolytica and has a sensitivity of approximately 87% and specificity greater than 90%. Polymerase chain reaction (PCR) assays on stool samples provide 90% to 95% sensitivity and 95% to 100% specificity. In selected cases, mucosal biopsy may be required to identify cysts or trophozoites. Serologic antibody tests are particularly useful in diagnosing extraintestinal disease, including liver abscess, although they may not distinguish recent from remote infection. Aspiration of abscess fluid is sometimes necessary to differentiate amebic from bacterial liver abscesses.
Imaging studies, including abdominal ultrasound or CT scanning, are essential for diagnosing amebic liver abscess and assessing its size, location, and potential complications.
Treatment is required for all patients infected with E. histolytica, regardless of symptom severity. Nonpathogenic Entamoeba species do not require therapy. Management of invasive disease typically begins with tinidazole, which is preferred for initial parasitic clearance, followed by an intraluminal amebicide such as paromomycin or diloxanide furoate to eradicate intestinal cysts. Metronidazole is an acceptable alternative to tinidazole for both intestinal disease and liver abscess. Amebic liver abscess generally responds well to medical therapy, but surgical intervention may be necessary in cases of toxic megacolon, impending rupture, or extension of the abscess into adjacent structures, including the pericardium.
Regarding disposition, host immunity after infection is incomplete, and reinfection rates are high in individuals who remain at risk. Referral to an infectious diseases specialist is recommended for patients with extraintestinal disease, persistent infection, or relapse. Surgical consultation is indicated for patients with toxic megacolon or complicated liver abscesses.
Important clinical considerations include the frequent coexistence of other intestinal parasites, particularly Giardia lamblia, and the high prevalence of nonpathogenic E. dispar colonization among homosexual men, which can complicate diagnosis. In the United States, E. histolytica remains the third most commonly isolated pathogen in returning travelers with infectious gastroenteritis, underscoring the importance of maintaining a high index of suspicion in appropriate clinical settings.