- Published on
KembaraXtra-Medicine – Amyloidosis
Amyloid is an altered, insoluble protein folded in β-pleated sheets that is deposited extracellularly. Amyloid can accumulate in one or many organs, causing organ dysfunction. Amyloidosis refers to a heterogeneous group of disorders characterized by deposition of an amorphous, extracellular fibrillar protein in various organs and tissues. Subtypes include primary amyloidosis (AL), secondary amyloidosis (AA), hereditary amyloidosis, and localized amyloidosis. ICD-10CM codes include E85.0 (non-neuropathic heredofamilial), E85.1 (neuropathic heredofamilial), E85.2 (heredofamilial, unspecified), E85.3 (secondary systemic), E85.4 (organ-limited), E85.8 (other), and E85.9 (unspecified).
Amyloidosis nomenclature is based on the amyloid protein precursor and associated clinical features. AL or AH derives from immunoglobulin light or heavy chain and is primary or localized, often associated with myeloma or macroglobulinemia. AA derives from serum amyloid A (SAA) and occurs as secondary amyloidosis or with familial Mediterranean fever and other familial periodic fever syndromes. ATTR derives from transthyretin and is familial or senile. A fibrinogen derives from fibrinogen and is associated with familial renal amyloidosis (Ostertag type). Aβ2M derives from β2-microglobulin and is dialysis-associated, including carpal tunnel syndrome. Aβ derives from amyloid-β precursor protein (ABPP) and is associated with Alzheimer disease. Apo A-I/A-II–related forms include apolipoprotein A-I (proteinuria) and apolipoprotein A-II (cardiac and neuropathy patterns). A lysozyme involves lysozyme deposition affecting the GI tract, liver, and kidneys. ALECT2 is noted as a renal form (leukocyte chemotactic factor 2 amyloidosis).
A major classification system (not exhaustive) includes: AA (serum amyloid A) causing systemic amyloidosis, often predominantly renal, linked to acquired or hereditary chronic inflammatory disease (formerly “secondary/reactive”); AL (monoclonal immunoglobulin light chains) causing systemic amyloidosis potentially involving many organs and associated with myeloma/monoclonal gammopathy/occult B-cell dyscrasias (formerly “primary”); and ATTR wild-type (normal plasma transthyretin) causing nonhereditary systemic amyloidosis with predominantly cardiac involvement (formerly “senile cardiac amyloidosis”). ATTR variant (genetic variants such as ATTR Met30, Ala60, Ile122) can cause familial amyloid polyneuropathy (FAP) often with prominent cardiomyopathy, and certain mutations (e.g., TTR Ile122) may produce predominantly cardiac involvement without neuropathy. Aβ2M (β2-microglobulin) causes dialysis-related amyloidosis with renal failure and long-term dialysis, with predominantly articular/periarticular involvement. Aβ causes cerebrovascular and intracerebral plaque amyloid in Alzheimer disease and may be familial. AApoAI (genetic variants of apolipoprotein AI) causes autosomal dominant systemic amyloidosis, typically nonneuropathic with visceral involvement—especially nephropathy—and minor wild-type deposits can occur in the aorta with aging. AApoAII causes autosomal dominant systemic amyloidosis with predominantly renal involvement. AFib (fibrinogen A α-chain variants) causes autosomal dominant systemic amyloidosis with nonneuropathic predominant nephropathy. ALys (lysozyme variants) causes autosomal dominant systemic amyloidosis with predominantly renal and GI involvement and can rarely present with hepatic rupture. ACys (cystatin C variant) is linked to hereditary cerebral hemorrhage with cerebral and systemic amyloidosis in Icelandic individuals. AGel (gelsolin variants) causes autosomal dominant systemic amyloidosis with predominantly cranial nerve involvement plus lattice corneal dystrophy and is described as most common in Finland.
In the United States, incidence is estimated at 1500–3500 new cases annually, with AL being the most common type. Amyloidosis primarily affects men ages 60–70 years. Common presenting symptoms include fatigue, dyspnea, edema, paresthesias, and weight loss, and other findings depend on organ involvement. Diagnosis should be considered in patients with a multisystem disorder involving the heart, kidney, liver, or nervous system. Renal involvement may present with nephrotic syndrome. Pulmonary involvement may cause fatigue and dyspnea. GI involvement is uncommon but may present with diarrhea, nausea, abdominal pain, and macroglossia. Cardiac involvement produces an infiltrative cardiomyopathy with preserved ejection fraction and diastolic dysfunction; in transthyretin-related disease, transthyretin dissociation leads to deposition of misfolded monomers as amyloid fibrils in the myocardium with subsequent cardiomyopathy and heart failure. Patients may have bleeding problems from factor X deficiency or fragile vessels infiltrated by amyloid; periorbital bleeding (“raccoon eyes”) is characteristic. Nervous system involvement can cause peripheral neuropathy, tendinopathy, muscle weakness, numbness, syncope, or dizziness, and associated autonomic neuropathy can be severely disabling. Syndromes in primary (AL) amyloidosis include nephrotic/nephrotic with renal failure (30%), hepatomegaly (24%), congestive heart failure (22%), carpal tunnel (21%), neuropathy (17%), and orthostatic hypotension (12%).
The common mechanism is deposition of Congo red–staining amyloid fibrils, but key subtype differences exist. AL is associated with an underlying clonal plasma cell disorder producing abnormal light chains with possible multi-organ deposition. AA has no plasma cell disorder and results from long-standing systemic inflammation (examples listed include tuberculosis, leprosy, malaria, untreated syphilis). Localized amyloidosis results from localized fibril synthesis without a plasma cell disorder. Familial amyloidosis includes forms due to transthyretin gene (TTR) mutations, with hepatocyte-derived transthyretin deposition in peripheral nerves and the heart.
Differential diagnosis depends on organ involvement. For renal disease consider toxin- or drug-induced necrosis, glomerulonephritis, renal vein thrombosis. For pulmonary disease consider sarcoidosis, connective tissue disease, infectious causes. For restrictive cardiomyopathy consider endomyocardial fibrosis or viral myocarditis. For carpal tunnel consider rheumatoid arthritis, hypothyroidism, overuse. For peripheral neuropathy consider alcohol abuse, vitamin deficiencies, diabetes mellitus.
Workup includes blood and urine testing for abnormal light chains, tests for target-organ damage, and histologic confirmation. Typical confirmation is via fat pad aspiration and bone marrow biopsy with Congo red staining. Recommended diagnostic evaluation includes CBC; chemistries and other labs (sodium, potassium, alkaline phosphatase, calcium, phosphorus, AST, bilirubin, creatinine, β2-microglobulin, glucose, cholesterol, uric acid, thyroid profile); serum and urine immunofixation; assays for immunoglobulin free light chains and immunoglobulins G, A, and M; cardiac markers (troponin, NT-proBNP); coagulation evaluation (factor X and prothrombin time); and cardiac/pulmonary assessment with chest x-ray, ECG, and echocardiogram with Doppler and strain imaging. Amyloid staging assigns 1 point each for FLC-diff ≥18 mg/dL, cTnT ≥0.025 ng/mL, and NT-proBNP ≥1800 pg/mL, producing stages I–IV (scores 0–3). Reported median survival (months) by stage is 94.1, 40.3, 14, and 5.8, respectively. Laboratory testing also includes SPEP/UPEP with immunofixation for light chains, routine labs such as BUN/creatinine, liver function tests, thyroid function, urine albumin, and biopsy confirmation; if fat pad biopsy is negative, biopsy of the affected organ may be required.
Imaging can help define organ involvement. Cardiac MRI may show a distinctive pattern. Two-dimensional Doppler echocardiography is useful for cardiac evaluation but is less sensitive than cardiac MRI. Nuclear imaging with technetium-labeled aprotinin may detect cardiac amyloidosis. Labeled diphosphonates are important for typing amyloidosis and diagnosing heart involvement in transthyretin cardiac amyloidosis, and bone scintigraphy may detect cardiac involvement earlier than echocardiography in transthyretin patients. Serum amyloid P (SAP) scintigraphy has high sensitivity for detecting deposits in liver, spleen, kidneys, adrenal glands, bones.
Acute/general management and disease-directed therapy depend on subtype and organ involvement. Patients with AL amyloidosis should be evaluated for autologous hematopoietic stem cell transplantation (HSCT); good candidates include those with good performance status, age <70, and limited organ involvement. tafamidis is an oral transthyretin stabilizer fda approved for adults with transthyretin-mediated amyloid cardiomyopathy (attr-cm) has been shown to decrease hospitalizations deaths in attr-cm. al, chemotherapy aims reduce production of amyloidogenic light chains by targeting clonal plasma cells; agents used multiple myeloma are effective, including melphalan prednisone, imids (thalidomide, lenalidomide) proteasome inhibitors also used. patients ineligible autologous hsct should receive melphalan- or bortezomib-based regimens. trials oligonucleotide drugs (inotersen, patisiran) have improvement clinical manifestations hereditary amyloidosis. who develop renal failure may require hemodialysis transplant. liver transplantation successfully familial secondary (aa) amyloidosis, recognition treatment the underlying disorder needed. newly diagnosed al show that adding daratumumab bortezomib, cyclophosphamide, dexamethasone associated higher hematologic complete response rates.< />pan>
Major treatment options include, for AL amyloidosis, IV melphalan with autologous stem cell rescue (G-CSF mobilized peripheral blood stem cell collection, IV melphalan 140–200 mg/m², autologous stem cell reinfusion), cyclic oral melphalan and dexamethasone (melphalan 0.22 mg/kg/day ×4 days; dexamethasone 20–40 mg/day ×4 days or weekly; repeat every 4 weeks), immunomodulators (lenalidomide 5–15 mg/day ×21 days with dexamethasone 20–40 mg weekly; repeat every 4 weeks), and proteasome inhibitors (IV bortezomib 0.7–1.6 mg/m² 1–2 times per week; repeat every 3–5 weeks). For AA amyloidosis, options include aggressive treatment of underlying inflammatory disease, medical or surgical treatment of infection, colchicine 1.2–1.8 mg/day for AA secondary to familial Mediterranean fever, and investigational antifibril drug eprodisate. For ATTR, options include orthotopic liver transplantation, transthyretin stabilizers (tafamidis, investigational diflunisal), and investigational oligonucleotide drugs (inotersen, patisiran).
Supportive treatment across all amyloidosis types is symptom- and organ-directed. For cardiac congestive failure, options include salt restriction 1–2 g/day and diuretics (furosemide, spironolactone, metolazone). For arrhythmias, consider pacemaker, automatic implantable cardiac defibrillator, and antiarrhythmics. For renal nephrotic syndrome, use salt restriction 1–2 g/day, elastic stockings and leg elevation, maintaining dietary protein, and an ACE inhibitor if blood pressure tolerates. For renal failure, use dialysis (long-term ambulatory peritoneal dialysis or hemodialysis). For autonomic nervous system orthostatic hypotension, options include midodrine, increased dietary salt or fludrocortisone depending on edema, and elastic stockings. For gastric atony/ileus, use small frequent low-fat feedings (6/day), oral nutritional supplements, jejunostomy tube feeding, or parenteral nutrition. For GI diarrhea, options include a low-fat diet (≤40 g), psyllium, loperamide, tincture of opium, and parenteral nutrition. For macroglossia, use a soft solid diet; partial glossectomy is rarely effective. For peripheral sensory neuropathy, avoid trauma and use medications such as gabapentin 100–300 mg three times daily, amitriptyline 25–50 mg at bedtime, or pregabalin 50–100 mg three times daily. For motor neuropathy with footdrop, use ankle-foot orthotics and physical therapy. For hematologic issues with intracutaneous bleeding, avoid trauma and antiplatelet agents; for factor X deficiency, use factor replacement (recombinant factor VIIa, prothrombin complex concentrates). Splenectomy may be considered for splenomegaly.
Prognosis is determined primarily by cardiac involvement and the amyloidosis form. In endomyocardial biopsy–documented cardiac amyloidosis, longer-term survival correlates more strongly with NYHA functional class than with ECG or echocardiographic variables. In AA amyloidosis, eradication of the predisposing disease slows and can occasionally reverse progression, with a median survival after diagnosis of 133 months. Patients with familial amyloidotic polyneuropathy often have a prolonged course of 10–15 years. Dialysis-associated amyloidosis progression can be improved with newer dialysis membranes that pass β2-microglobulin. Median survival in patients with overt congestive heart failure is approximately 6 months, compared with 30 months without CHF.

- Published on
KembaraXtra-Medicine – Amphetamine Use Disorder
Amphetamine use disorder is a pattern of amphetamine use marked by impairment in control of use or social obligations, risky use, or physical dependence within a 12-month period. Severity is categorized as mild, moderate, or severe based on the number of symptoms. Amphetamines are central nervous system stimulants that increase monoamine neurotransmitters (dopamine, norepinephrine, serotonin) and are defined by a phenylethylamine structure. Amphetamine and dextroamphetamine are FDA approved for AD/HD, narcolepsy, and obesity. Methamphetamine is a type of amphetamine that is FDA approved for AD/HD and obesity, but is more commonly illicitly produced and used recreationally. Synonyms include stimulant use disorder and methamphetamine use disorder. ICD-10CM codes include F15.10 (mild), F15.20 (moderate or severe), F15.11 (mild, in early or sustained remission), and F15.21 (moderate or severe, in early or sustained remission).
Epidemiologically, prevalence is described as rare: 0.6% among people ages ≥12 years (about 1.5 million) with methamphetamine use disorder, and 0.3% among people ≥12 years (about 758,000) with prescription stimulant use disorder. Among adults who used methamphetamine in the past year, 53% met criteria for methamphetamine use disorder. Methamphetamine use disorder is more common among those age 26 and older (0.6%) than under age 25, while prescription stimulant use disorder is more common between ages 12 and 25 compared with ≥26.
Risk increases in people with any substance use disorder. Among psychiatric comorbidities, bipolar disorder, schizophrenia, and antisocial personality disorder are associated with stimulant use disorder. Demographic risk factors for methamphetamine use include male sex, lower educational attainment, and non-Hispanic White ethnicity. Methamphetamine use is higher among men who have sex with men, including in “chemsex,” where methamphetamine and other stimulants are used to increase arousal and reduce inhibition during sexual sessions.
Clinically, amphetamine use disorder can develop as quickly as within 1 week of exposure, and most people experience physical dependence in the form of tolerance or withdrawal. Amphetamines may be used by oral ingestion, insufflation, injection, or smoking, and use is often described as chronic or episodic binges. During intoxication, patients may report euphoria and may present with hyperarousal, aggressive behavior, psychosis (paranoia and hallucinations), panic attacks, or suicidal and homicidal ideation. With toxicity, sympathomimetic signs such as hypertension, tachycardia, dilated pupils, and flushed skin can occur. Some intoxicated patients report chest pain, and amphetamine use increases risk of myocardial infarction, arrhythmias, and strokes. During withdrawal, patients commonly report depressive symptoms. Risk for psychosis increases with higher frequency and amount of use, and psychosis can occur within months to several years after starting amphetamine use.
The exact cause of amphetamine use disorder is not known, but amphetamine-type medications act on the dopamine and monoamine transporter (DAT and VMAT2), increasing dopamine transmission in the mesolimbic dopamine pathway (the reward pathway).
In diagnosis, amphetamine use disorder is different from amphetamine misuse, which refers to use inconsistent with prescription guidelines. Patients using PCP or novel psychoactive substances (such as cathinones/bath salts) may present similarly and can be distinguished with a toxicology test. Primary psychiatric disorders (e.g., schizophrenia) or depressive disorders (e.g., major depressive disorder) can be evaluated by establishing a timeline of symptoms relative to amphetamine use; psychosis that quickly improves is more consistent with amphetamine-induced psychosis.
Work-up may be difficult because detailed history can be hard to obtain during acute intoxication or withdrawal. An ECG should be performed in patients with tachycardia. A safety assessment is essential to determine whether the patient can safely care for themselves, especially when suicidal or homicidal ideation is reported. Recommended labs include basic testing such as Chem 7, serum lactate, creatine phosphokinase, thyroid function tests, and hepatic enzymes. A urine or serum toxicology test can confirm amphetamine presence. There are no radiographic tests for diagnosis of stimulant use disorder, but CT imaging may be considered for strokes or vasculopathies associated with amphetamine use.
Treatment is primarily nonpharmacologic, with behavioral interventions as the mainstay. Approaches such as contingency management, cognitive-behavioral therapy, behavioral activation, and exercise have shown mild effectiveness in reducing symptoms and sustaining abstinence. Contingency management, especially when combined with community reinforcement (reorganizing the environment so abstinence is more rewarding than drug use), has been particularly shown to be effective.
For acute general management, antipsychotics—especially olanzapine and haloperidol—have evidence for treating amphetamine-associated psychosis. No medication has been shown to be effective for amphetamine withdrawal. There is limited evidence that amineptine (a tricyclic antidepressant no longer available due to abuse potential) reduced discontinuation symptoms and improved clinical presentation, and there is mixed evidence that mirtazapine may reduce anxiety symptoms during withdrawal.
For chronic management, there is no evidence that pharmacologic treatment alone is sufficient. Randomized controlled trials show limited benefit from methylphenidate, bupropion, modafinil, and naltrexone in reducing amphetamine use.
Disposition-wise, methamphetamine use disorder often follows a course of intense use, sobriety, and relapse. Complications of amphetamine intoxication (including ecstasy) include hyponatremia due to SIADH (very rare; managed with water restriction), intracerebral hemorrhage (uncommon; CT/MRI if focal signs or reduced GCS), serotonin toxicity (uncommon; supportive care and cyproheptadine for serious cases), hypertension (uncommon; GTN infusion, benzodiazepines, labetalol; avoid beta-blockers), hyperthermia (common; cooling measures, diazepam, cyproheptadine), rhabdomyolysis (uncommon; IV fluids and cooling), supraventricular and ventricular tachycardia (uncommon; short-acting antiarrhythmics such as esmolol for SVT), and seizures (uncommon; IV diazepam).
Referral is typically to physicians and mental health professionals. Important pearls include that stimulant use disorders often co-occur with sedative substance use disorders (e.g., alcohol, marijuana, opioids) because these may relieve stimulant side effects such as insomnia and anxiety. Amphetamine use is also associated with an increased risk of death by suicide, so assessing suicidality is critical in any patient presenting with amphetamine use.
For prevention, community-based universal prevention programs that promote youth social-emotional skills and reduce risk factors have been shown to lead to sustained reductions in lifetime methamphetamine use.

- Published on
KembaraXtra- Medicine – Primary Aldosteronism (Hyperaldosteronism)
Primary aldosteronism is a group of disorders characterized by autonomous and excessive secretion of aldosterone, leading to renal sodium retention, suppression of plasma renin, and secondary hypertension. The condition classically presents with hypertension, hypokalemia, and metabolic alkalosis, although many patients may be normokalemic. Because aldosterone excess directly affects cardiovascular and renal systems, primary aldosteronism is now recognized as a common and clinically important cause of secondary hypertension.
Epidemiologic studies show that primary aldosteronism is far more prevalent than previously believed, particularly among patients with resistant hypertension. Current estimates suggest it may be present in 17% to nearly 27% of patients with resistant hypertension, with prevalence increasing as the severity of hypertension worsens. Familial forms of hyperaldosteronism are uncommon but should be suspected in patients with early-onset hypertension (before age 21), a family history of primary aldosteronism, or a family history of stroke before age 40.
The most common causes of primary aldosteronism include bilateral idiopathic adrenal hyperplasia, which accounts for approximately 60% to 70% of cases, and unilateral aldosterone-producing adenomas (Conn syndrome), which account for about 30% to 40%. Less common causes include unilateral adrenal hyperplasia, familial hyperaldosteronism, adrenal carcinoma, and ectopic aldosterone production. The underlying cause is important because it directly determines treatment strategy.
Clinically, many patients are asymptomatic aside from hypertension. When present, manifestations may include resistant hypertension, spontaneous or diuretic-induced hypokalemia, muscle weakness, fatigue, or a history of early cardiovascular events. A family history of early hypertension or stroke should heighten suspicion. Because symptoms may be subtle or absent, a high index of suspicion is required, particularly in patients whose blood pressure remains uncontrolled despite multiple antihypertensive agents.
Diagnosis begins with recognition of the biochemical pattern of suppressed plasma renin activity and elevated serum aldosterone levels, typically with aldosterone concentrations exceeding 15 ng/dL. Screening is recommended in patients with resistant hypertension, early-onset hypertension, hypokalemia, adrenal incidentalomas, sleep apnea, or a strong family history of hypertension or stroke. The aldosterone-to-renin ratio (ARR) is commonly used as an initial screening test, although its sensitivity is limited. When results are inconclusive, 24-hour urinary aldosterone excretion under aldosterone-suppressing conditions may be required for confirmation.
Routine laboratory findings are not diagnostic but may support the diagnosis, including hypokalemia, metabolic alkalosis, and occasionally mild hypernatremia. Once biochemical confirmation is obtained, imaging is performed to determine whether aldosterone excess is unilateral or bilateral. Adrenal CT scanning is the preferred imaging modality, as adrenal adenomas are typically lipid-rich and well visualized on CT. MRI may be used as an alternative. However, imaging alone cannot reliably distinguish unilateral from bilateral disease.
To definitively differentiate unilateral from bilateral aldosterone secretion, adrenal vein sampling (AVS) is recommended in most patients who are surgical candidates. This distinction is critical, as unilateral disease is best treated surgically, whereas bilateral disease requires medical therapy. AVS should be performed by experienced interventional radiologists to ensure accurate results.
Management includes both nonpharmacologic and pharmacologic strategies. Lifestyle measures such as regular blood pressure monitoring, a low-sodium diet, tobacco avoidance, weight management, and regular exercise are recommended for all patients. Acute management focuses on controlling hypertension and correcting hypokalemia, typically using mineralocorticoid receptor antagonists such as spironolactone or eplerenone, or potassium-sparing agents like amiloride.
Long-term medical therapy is the mainstay for patients with bilateral adrenal hyperplasia. Spironolactone is usually initiated at a low dose and titrated upward every two weeks to achieve a normal serum potassium level without supplementation. Eplerenone is an effective alternative with fewer antiandrogenic side effects, such as gynecomastia or menstrual irregularities. Amiloride may be used in patients who cannot tolerate mineralocorticoid receptor antagonists.
For patients with unilateral aldosterone-producing adenomas or unilateral adrenal hyperplasia, laparoscopic adrenalectomy is the treatment of choice. Surgical treatment typically resolves hypokalemia and improves blood pressure control, although hypertension persists in 40% to 65% of patients postoperatively. Complete resolution of hypertension is more likely in younger patients, those with a shorter duration of hypertension, fewer preoperative antihypertensive medications, no family history of hypertension, and higher aldosterone-to-renin ratios.
Overall, treatment of primary aldosteronism—whether surgical or medical—results in meaningful improvements in blood pressure control, potassium balance, and cardiovascular and renal outcomes. Untreated disease is associated with higher risks of stroke, myocardial infarction, atrial fibrillation, left ventricular hypertrophy, and decline in kidney function compared with essential hypertension. These risks are significantly reduced with appropriate therapy, and in many cases, surgical treatment lowers cardiovascular risk to below that of matched patients with essential hypertension.

- Published on
KembaraXtra-Medicine – Amenorrhea
Amenorrhea means the absence of menstruation. It is classified as primary or secondary depending on whether the patient has had previous menstrual cycles. A workup for primary amenorrhea is indicated for any adolescent age 15 or older who has not reached menarche, or who does not reach menarche within 3 years of thelarche. Secondary amenorrhea is defined as the absence of menses for more than 6 months in a patient who previously had normal menstrual progesterone withdrawal cycles. ICD-10CM codes include N91.0 (Primary amenorrhea), N91.1 (Secondary amenorrhea), and N91.2 (Amenorrhea, unspecified).
From an epidemiologic standpoint, the incidence in the United States is <1% for primary amenorrhea and 5% to 7% secondary amenorrhea, with no racial or ethnic predilection.< />pan>
Amenorrhea can be physiologic or pathologic. Physiologic causes include pregnancy, lactation, constitutional delay of puberty, and menopause. Pathologic causes differ by whether amenorrhea is primary or secondary. For primary amenorrhea, major categories include hypergonadotropic hypogonadism (including 45,X, 46,XX, 46,XY, and 46,XX with gonadal failure), eugonadism (including Müllerian agenesis, vaginal septum, imperforate hymen, polycystic ovarian syndrome, congenital adrenal hyperplasia, Cushing disease, thyroid disease, and androgen insensitivity), and hypogonadotropic hypogonadism (including constitutional delay, GnRH deficiency, and other CNS disease). For secondary amenorrhea, common causes include PCOS (about 28%), eating disorders/stress (about 15.5%), hypothyroidism (about 1.5%), Sheehan syndrome (about 1.5%), Cushing syndrome (about 1%), pituitary tumor/empty sella (about 2%), pituitary diseases (about 5%), eating disorders/stress (about 2%), nonclassic congenital adrenal hyperplasia (about 0.5%), ovarian tumor (about 1%), hyperprolactinemia (about 13%), and anatomic causes (about 7%, including Asherman syndrome). An abnormal karyotype is also listed as a possible contributor in secondary amenorrhea (0.5%).
Among congenital anatomic causes of primary amenorrhea with normal breast development (cervix not visible on pelvic exam; short vagina that may be absent or obstructed), key diagnoses include Müllerian agenesis, androgen insensitivity (AI), transverse vaginal septum, and imperforate hymen. These account for about 15%, 1%, 3%, and 1% of primary amenorrhea cases, respectively; among those with apparent obstruction or absence of the vagina, proportions are approximately 75%, 5%, 15%, and 5%, respectively. Chromosomes are 46,XX for Müllerian agenesis, transverse vaginal septum, and imperforate hymen, and 46,XY for androgen insensitivity. Gonads are ovaries in Müllerian agenesis, transverse vaginal septum, and imperforate hymen, and testes in androgen insensitivity. Serum testosterone is at normal female levels in Müllerian agenesis, transverse vaginal septum, and imperforate hymen, and at normal male (high) levels in androgen insensitivity. The vagina is absent or shallow in Müllerian agenesis and androgen insensitivity, obstructed by a septum (thick or thin; high or low) in transverse vaginal septum, and obstructed by a thin membrane (sometimes bluish from hematocolpos) in imperforate hymen. Axillary/pubic hair is present in Müllerian agenesis, transverse vaginal septum, and imperforate hymen, but absent in complete androgen insensitivity (unless incomplete). Cyclic pain may be variable in Müllerian agenesis, absent in androgen insensitivity, and present in transverse vaginal septum and imperforate hymen. The uterus is absent or rudimentary in Müllerian agenesis, absent in androgen insensitivity, and present in transverse vaginal septum and imperforate hymen. A mass is typically absent in Müllerian agenesis and androgen insensitivity, but may be present in transverse vaginal septum and imperforate hymen and can cause acute urinary retention due to hematocolpos obstructing the urethra; with imperforate hymen, the introitus may bulge with Valsalva. Associated anomalies include urinary tract and skeletal anomalies in Müllerian agenesis, inguinal hernias and risk of gonadal malignancy in adulthood in androgen insensitivity, major urinary tract abnormalities in ~15% with transverse vaginal septum, and possibly some increased urinary tract abnormalities in imperforate hymen. Treatment includes vaginal dilation or surgical neovagina for Müllerian agenesis and transverse vaginal septum, gonadectomy after age 16–18 for androgen insensitivity, surgery tailored to septum extent/location (often as soon as possible) for transverse vaginal septum, and excision of hymen as soon as possible for imperforate hymen (diagnostic needle aspiration is contraindicated due to infection risk). Fertility usually requires advanced reproductive technology (including IVF with a surrogate uterus) in Müllerian agenesis, is not possible in androgen insensitivity, is variable in transverse vaginal septum (low septa have better prognosis than high septa), and is usually preserved in imperforate hymen.
Clinical presentation varies by etiology. Turner syndrome usually presents with primary amenorrhea (unless mosaic) and may include short stature, epicanthic folds, low-set ears, high-arched palate, micrognathia, sensorineural hearing loss, otitis media, webbed neck, pigmented nevi, square/shield chest, widely spaced nipples, absent breast development, bicuspid aortic valve, coarctation of the aorta, cubit valgus, short fourth metacarpal, hyperconvex nails, leg edema, renal abnormalities, and autoimmune/metabolic issues including thyroiditis and diabetes mellitus. Pure gonadal dysgenesis causes amenorrhea but lacks Turner’s dysmorphic features. Müllerian agenesis is typically sporadic and presents with primary amenorrhea, normal breast development, normal pubic/axillary hair, normal female external genitalia, absent uterus and upper vagina, and present ovaries; some patients have renal and vertebral anomalies, and renal agenesis should be evaluated. Transverse vaginal septum and imperforate hymen present with primary amenorrhea, progressive cyclic lower abdominal pain, exam findings of septum/hymen abnormality, and perirectal fullness from hematocolpos. Androgen insensitivity syndrome presents with primary amenorrhea, may be X-linked recessive in some, with normal breast development, absent pubic/axillary hair, possible testes in groin/inguinal canal, and absent uterus and vagina, without renal/vertebral anomalies. Adult-onset congenital adrenal hyperplasia is more frequent in Ashkenazi Jewish, Inuit Native American, French Canadian, and Mexican populations, mimics PCOS, causes hyperandrogenism (virilization, hirsutism, acne), and can be associated with hypertension. 5-alpha reductase deficiency may cause primary amenorrhea with virilization at puberty. PCOS often presents with secondary amenorrhea/oligomenorrhea and is diagnosed by at least 2 of 3 Rotterdam criteria (oligo/anovulation, hyperandrogenism, polycystic ovaries on ultrasound defined by ≥12 follicles 2-9 mm or ovarian volume >10 cm³); it is associated with metabolic syndrome/obesity (60%–80%), insulin resistance, and predisposition to type 2 diabetes. Cushing syndrome (rare; prevalence about 1/1,000,000) can cause secondary amenorrhea, hyperandrogenism, dorsocervical fat pad, central obesity with thin extremities, abdominal striae, easy bruising, hypertension, and proximal muscle weakness. Hypothyroidism can cause secondary amenorrhea with lethargy, constipation, decreased appetite, weight gain, cold intolerance, hair loss, dry skin, and bradycardia. Primary ovarian insufficiency (previously premature ovarian failure) presents with secondary amenorrhea before age 40 with elevated FSH/LH, possible history of oophorectomy/radiation/chemotherapy, vasomotor symptoms, and thin dry vaginal mucosa without rugosity, and may be associated with autoimmune/karyotypic abnormalities. Hyperprolactinemia commonly causes secondary amenorrhea and may be medication-related (antipsychotics, OCPs, antidepressants, antihypertensives, H2 blockers, opioids, etc.) or due to pituitary adenoma (headache, vomiting, vision changes), and may present with galactorrhea. Sheehan syndrome follows postpartum hemorrhage with secondary amenorrhea, failure of lactation, and other hypopituitarism features. Asherman syndrome is associated with a history of D&C, secondary amenorrhea, and recurrent miscarriage/infertility. Functional hypothalamic disorders cause secondary amenorrhea associated with eating disorders, excessive exercise, stress, or street drug use. Kallmann syndrome is associated with anosmia due to developmental defects of GnRH neurons and the olfactory placode.
Diagnosis begins with the first step: rule out pregnancy using a serum or urine pregnancy test. Further evaluation depends on history and physical exam, including assessment of breast Tanner stage and whether a uterus is present, since the combination of present breasts with absent uterus narrows the differential primarily to Müllerian agenesis or androgen insensitivity syndrome. In primary amenorrhea, pelvic ultrasound or MRI is used to detect anatomic abnormalities of the uterus, cervix, ovaries, or vagina. Karyotyping is performed when the uterus is absent or Turner syndrome is suspected (e.g., 46,XX in Müllerian agenesis, 46,XY in AIS, 45,XO in Turner syndrome). Recommended labs include FSH, TSH/FT4, prolactin, and estradiol; an FSH ~40 mIU/mL with estradiol <20 pg />L suggests primary ovarian insufficiency, and prolactin >200 ng/mL suggests prolactinoma (noting thresholds vary by lab). Serum testosterone is checked when the uterus is absent or hyperandrogenism is present (male-range in AIS; female-range in Müllerian agenesis). 17-alpha hydroxyprogesterone is checked when hyperandrogenism is present to rule out CAH; these patients may also have elevated progesterone and deoxycorticosterone, hypernatremia, and hypokalemia. MRI of the head is obtained in the presence of hyperprolactinemia, visual field defects, or signs of hypothalamic–pituitary dysfunction.
In secondary amenorrhea, evaluation includes FSH, TSH/FT4, prolactin, and estradiol; low FSH with low estradiol indicates hypogonadotropic hypogonadism, and high FSH with low estradiol indicates hypergonadotropic hypogonadism. A progesterone withdrawal test is performed by giving medroxyprogesterone 10 mg for 10 days; withdrawal bleeding suggests euestrogenic anovulation with normal outflow tract and ovarian function. If no bleeding occurs, an estrogen-progesterone withdrawal is done using 25-35 days of estrogen (Premarin 0.625-2.5 mg daily) followed by 10 days of medroxyprogesterone; withdrawal bleeding indicates hypogonadism, while lack of bleeding suggests an end-organ/outflow tract problem such as Asherman syndrome. When hyperandrogenism is present, additional labs include LH, testosterone, and DHEA-S; testosterone >200 ng/mL raises concern for androgen-producing ovarian/adrenal tumors (may be mildly elevated in PCOS), and DHEA-S >700 mcg/dL suggests an adrenal source (again, moderate elevations warrant high suspicion and thresholds are not required when a tumor is evident). An LH/FSH ratio >2 can be seen in PCOS but is not part of Rotterdam diagnostic criteria. Imaging is tailored to suspected cause: pelvic ultrasound for PCOS or ovarian tumor, abdominal CT for adrenal tumor, head MRI when indicated, and hysterosalpingography, sonohysterography, or diagnostic hysteroscopy when Asherman syndrome is suspected. Rare additional tests include serum transferrin (hemochromatosis), serum ACE (sarcoidosis), and karyotyping when POI occurs before age 40.
Treatment depends on the underlying cause and the patient’s goals (e.g., treating hirsutism or achieving pregnancy). Patients with hypogonadism should receive estrogen replacement plus calcium and vitamin D to prevent osteoporosis; those with a uterus also need continuous or intermittent progesterone to prevent endometrial hyperplasia or cancer, often using combined oral contraceptives. Many anatomic abnormalities require surgical correction; creation of a neovagina for Müllerian agenesis is usually delayed until emotional maturity and readiness for dilation or postoperative care, and if correction is not feasible, pregnancy may require a surrogate. Associated urogenital anomalies (e.g., renal agenesis) must be assessed and treated appropriately. In androgen insensitivity syndrome, gonadal malignancy risk is about 22% but rarely occurs before age 20; laparoscopic gonadectomy is performed after breast development and adult stature, and because there is no uterus, patients require estrogen only (no progesterone). Adult-onset CAH may be treated with low-dose corticosteroids plus sex steroids to reduce ACTH-driven androgen excess. Primary ovarian insufficiency requires estrogen and progesterone replacement, and conception typically requires IVF using donor oocytes; these patients have increased risk of osteoporosis and heart disease and may have autoimmune associations (hypothyroidism, Addison disease, diabetes), so fasting glucose, TSH, and (when appropriate) morning cortisol should be checked. If a Y chromosome is present, gonadal tissue removal is recommended at diagnosis to reduce tumor risk. Hypothyroidism is treated with thyroid replacement. Hyperprolactinemia is treated by stopping culprit drugs when possible and/or using dopamine agonists such as bromocriptine or cabergoline; surgery for pituitary adenomas is uncommon but may be needed for visual compromise, resistance to medical therapy, or rapid growth. Hypothalamic amenorrhea is treated by addressing the cause (behavioral modification and nutritional counseling for eating disorders/excess exercise; estrogen therapy and osteoporosis prevention for those who cannot reduce training), and infertility can be treated with clomiphene citrate, exogenous gonadotropins, or pulsatile GnRH therapy. For PCOS, primary therapy is weight loss with diet and exercise, with additional options including OCPs or cyclic progestins for endometrial protection, metformin to improve insulin resistance and ovulatory function (noting mixed effectiveness in recent studies), OCPs and/or spironolactone for hyperandrogenism, and ovulation induction with clomiphene citrate or letrozole. Asherman syndrome is treated with hysteroscopic lysis of adhesions followed by long-term exogenous estrogen to promote endometrial regrowth. A genetics consultation is appropriate for hereditary causes, and psychiatry consultation may be required for major depression, anorexia nervosa, bulimia nervosa, or other major psychiatric disorders.
Complications of amenorrhea include osteoporosis, endometrial hyperplasia and uterine cancer, and infertility. Prognosis depends on the underlying cause.
Patient and family education should include reassurance that amenorrhea itself is not automatically dangerous, but patients with an intact endometrium must understand the risks of unopposed estrogen (whether exogenous hormone therapy or endogenous estrogen exposure as in PCOS). Hypoestrogenic patients should be counseled on the importance of estrogen replacement to prevent bone loss, and the patient’s future fertility goals and potential for childbearing should be discussed.
I
- Published on
KembaraXtra-Medicine – Amaurosis Fugax
Amaurosis fugax is defined as a transient loss of monocular vision caused by temporary retinal ischemia resulting from a reversible occlusion of the ophthalmic artery or its branches. The ophthalmic artery arises from the internal carotid artery, and the central retinal artery is a branch of the ophthalmic artery. This condition is classified under ICD-10CM code G45.3 and is considered an important warning sign of underlying vascular disease.
From an epidemiologic perspective, amaurosis fugax is an uncommon but clinically significant presentation of carotid artery disease in the United States. The peak incidence occurs around 55 years of age, and its recognition is critical because it often represents a form of transient ischemic attack (TIA) with potential risk for future stroke.
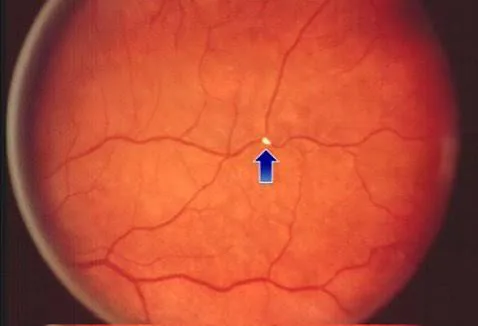
The clinical presentation is typically sudden in onset and lasts seconds to minutes. Patients often describe the vision loss as a shade or curtain descending over the eye, most commonly from above downward. The visual deficit may be complete or partial and can involve hemianopic or quadrantic vision loss. During the acute stage, cholesterol emboli, known as Hollenhorst plaques, may sometimes be visualized within the retinal arteries on funduscopic examination.
The etiology of amaurosis fugax is most often embolic in nature. Emboli may originate from the internal carotid artery, usually in the setting of carotid atherosclerotic stenosis, or from the heart, with atrial fibrillation being the most common cardiac source. Carotid artery emboli are typically identified as stenosis on carotid imaging such as CT angiography, MR angiography, or carotid ultrasound. Other important causes include giant cell arteritis, which produces inflammatory occlusion of retinal arteries; hyperviscosity syndromes such as sickle cell disease; hypercoagulable states; and reversible cerebral vasoconstriction syndrome.
In terms of differential diagnosis, retinal migraine must be considered, as it differs by having a gradual onset of visual symptoms over 15 to 20 minutes rather than sudden loss. Transient visual obscurations associated with papilledema also differ, as they are typically brief (1–2 seconds), may be binocular, and are related to transient increases in intracranial pressure. If vision loss persists at the time of evaluation, the differential should be expanded to include arteritic or nonarteritic anterior ischemic optic neuropathy, central retinal artery occlusion, and branch retinal artery occlusion.
The diagnostic workup should prioritize identifying embolic sources, while giant cell arteritis must always be considered, particularly in older adults. A careful retinal examination may reveal an embolus, which can confirm the diagnosis. Auscultation for carotid bruits, examination of peripheral pulses, and assessment for temporal artery tenderness are essential. Patients should be questioned about symptoms suggestive of giant cell arteritis, including headache, scalp tenderness, fever, and jaw claudication. A focused neurologic examination is necessary to evaluate for signs of hemispheric stroke, such as contralateral weakness, sensory loss, or aphasia.
Laboratory evaluation includes a complete blood count along with erythrocyte sedimentation rate and C-reactive protein to assess for inflammation, particularly when giant cell arteritis is suspected. Additional studies include serum chemistries, lipid profile, hemoglobin A1C, cardiac enzymes, and electrocardiography. A hypercoagulable workup may be pursued selectively in younger patients or when the history suggests an underlying thrombophilic disorder.
Imaging studies are central to evaluation. CT or MR angiography of the head and neck is preferred to assess carotid and intracranial vasculature, with carotid ultrasound as an alternative when angiography is contraindicated. MRI of the brain with diffusion-weighted imaging is recommended to identify silent or symptomatic infarcts. Transthoracic echocardiography is used to screen for cardiac embolic sources, while transesophageal echocardiography provides greater sensitivity for detecting left atrial appendage thrombi, patent foramen ovale, ventricular mural thrombus, or aortic arch atheroma. Extended cardiac rhythm monitoring is advised when neither carotid disease nor giant cell arteritis explains the presentation.
Management begins with nonpharmacologic therapy, including dietary modification to reduce saturated fats and cholesterol, regular exercise, and cessation of tobacco and illicit drug use. These measures address the underlying vascular risk factors that contribute to embolic disease.
From an acute management standpoint, amaurosis fugax should be treated as a medical emergency, similar to other forms of TIA. Aspirin therapy should be initiated promptly. If giant cell arteritis is suspected, high-dose prednisone must be started immediately, and the patient should be referred urgently for temporal artery biopsy within 48 hours.
Chronic management focuses on secondary stroke prevention. Carotid endarterectomy is recommended for patients with carotid stenosis greater than 50%, and carotid stenting may be considered in patients who are poor surgical candidates. Aggressive management of vascular risk factors—including hypertension, hyperlipidemia with statin therapy, diabetes, and smoking cessation—is essential. Antiplatelet therapy is indicated for atherosclerotic disease or following carotid intervention, while anticoagulation is required when atrial fibrillation is identified as the etiology.
Regarding prognosis, patients with greater than 50% carotid stenosis who do not undergo carotid endarterectomy and present with transient monocular blindness have an estimated 10% risk of stroke within 3 years, compared with approximately 20% risk in those presenting with hemispheric TIA.
Appropriate referral is critical. As with any TIA, patients should undergo emergent inpatient evaluation at a certified stroke center whenever possible. Carotid endarterectomy or stenting should be considered in patients with ipsilateral high-grade stenosis (≥70%), and may also be appropriate for stenosis of 50%–69% or in patients experiencing multiple TIAs despite optimal medical therapy.
Key clinical pearls include recognizing that visualization of cholesterol emboli in retinal arteries on fundoscopy confirms the diagnosis, understanding that transient visual loss has multiple potential causes, and ensuring that amaurosis fugax is managed with the same urgency as other transient ischemic attacks.
- Published on
KembaraXtra-Medicine – Alzheimer Disease
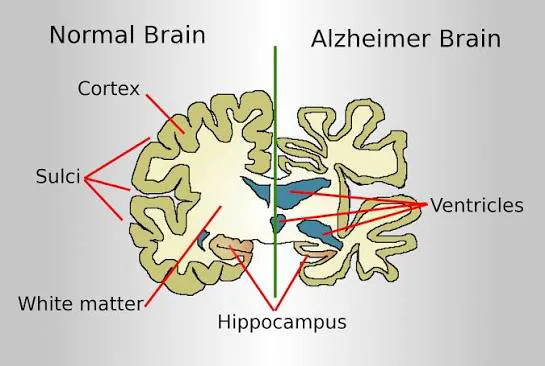
Dementia is a syndrome characterized by progressive loss of previously acquired cognitive skills, including memory, language, insight, and judgment. Alzheimer disease (AD) is thought to account for the majority of all cases of dementia. The ICD-10CM codes for Alzheimer disease include G30.0 (Alzheimer disease with early onset), G30.1 (Alzheimer disease with late onset), G30.8 (other Alzheimer disease), and G30.9 (Alzheimer disease, unspecified).
The incidence of Alzheimer disease increases markedly with age, with risk doubling every 5 years after age 65. The Chicago Health and Aging Population study demonstrated an average annual incidence of 2.3% among individuals aged 65 years and older, with Black individuals having a significantly increased risk compared with White individuals. In terms of prevalence, approximately 1 in 9 people (10.7%) aged 65 years or older has Alzheimer dementia. In the United States, an estimated 6.5 million people are affected, including 5% of individuals aged 65–74 years, 13.1% of those aged 75–84 years, and 33.2% of those aged 85 years or older. Additionally, 12%–18% of adults over age 60 are believed to have mild cognitive impairment (MCI). Alzheimer disease affects women more frequently than men, with approximately 4 million women and 2.5 million men affected nationwide.
Clinically, Alzheimer disease most often presents insidiously. A spouse or close family member usually first notices progressive memory impairment rather than the patient. Individuals have difficulty learning and retaining new information, managing complex tasks such as finances, and demonstrate impairments in judgment, reasoning, visuospatial abilities, and orientation, often leading to problems such as getting lost or difficulty driving. Behavioral and personality changes, including apathy and mood disturbances, commonly accompany memory loss, and later stages may involve agitation, delusions, or psychosis. Rare atypical variants include early and severe behavioral changes, focal neurologic findings, parkinsonism, hallucinations, frequent falls, or onset of symptoms at or before 65 years of age.
The diagnosis of Alzheimer disease has evolved with the development of biomarkers that demonstrate Alzheimer pathology in vivo, particularly amyloid beta deposition and pathologic tau accumulation. The National Institute on Aging–Alzheimer’s Association (NIA-AA) criteria, initially published in 2011 and revised in 2018, conceptualize Alzheimer disease as a biologic process rather than solely a clinical syndrome. These criteria define three stages: preclinical Alzheimer disease (biologic evidence without symptoms), MCI due to Alzheimer disease (mild memory impairment without functional decline but with biomarker evidence), and dementia due to Alzheimer disease (cognitive decline with functional impairment and biomarker confirmation). The 2018 framework further introduced the ATN system, defining disease by amyloid (A), tau pathology (T), and neurodegeneration (N). Although biomarkers are not yet routinely used in general clinical practice, they are integral to research and clinical trials.
In routine clinical settings, Alzheimer disease is typically diagnosed using clinical history, neurologic examination, and established criteria such as DSM-5 or NINCDS-ADRDA. Core features include progressive memory loss plus at least one additional cognitive deficit (aphasia, apraxia, agnosia, or executive dysfunction), decline in social or occupational functioning, insidious onset with gradual progression, objective documentation of cognitive loss, and exclusion of alternative causes. Red flags suggesting an alternative diagnosis include age younger than 65 years, fluctuating consciousness, rapid progression over months, prominent early behavioral changes, or focal neurologic findings such as gait disturbance, lateralized weakness, myoclonus, or parkinsonism.
The differential diagnosis of Alzheimer disease is broad and includes other neurodegenerative dementias such as frontotemporal lobar degeneration, dementia with Lewy bodies, Parkinson disease dementia, corticobasal syndrome, progressive supranuclear palsy, limbic-predominant age-related TDP-43 encephalopathy, and primary age-related tauopathy. Nondegenerative conditions include vascular cognitive impairment, depression (pseudodementia), subjective memory complaints, brain tumors, chronic subdural hematoma, infections such as HIV or neurosyphilis, toxic or metabolic encephalopathies, and organ failure–related cognitive impairment.
Evaluation begins with a comprehensive history and physical examination, including careful medication review to identify agents that impair cognition, such as anticholinergics, benzodiazepines, opioids, barbiturates, and neuroleptics. Screening for depression is essential, as it may mimic or coexist with dementia. Cognitive screening tools such as the Mini-Mental State Examination, Mini-Cog, and Montreal Cognitive Assessment are commonly used, while formal neuropsychologic testing is reserved for atypical cases or diagnostic uncertainty. In Alzheimer disease, episodic memory deficits typically appear early, whereas attention is often preserved until later stages.
Laboratory evaluation usually includes complete blood count, serum electrolytes, glucose, renal and liver function tests, thyroid function tests, and vitamin B12 levels, with additional testing guided by clinical context. Neuroimaging, preferably MRI, is used to exclude alternative diagnoses and assess characteristic patterns of neurodegeneration, such as hippocampal atrophy. Amyloid and tau PET imaging may support diagnosis in selected cases but are not diagnostic on their own.
Management of Alzheimer disease incorporates both nonpharmacologic and pharmacologic strategies. Nonpharmacologic care focuses on patient safety, caregiver education, behavioral interventions, and cognitive stimulation. Addressing driving safety, wandering, medication management, and household hazards is essential. Behavioral symptoms such as agitation, repetitive questioning, and social withdrawal often respond to structured routines and environmental modifications. A person-centered, interdisciplinary approach tailored to disease stage improves quality of life for both patients and caregivers.
Pharmacologic treatment is primarily symptomatic. Cholinesterase inhibitors (donepezil, rivastigmine, and galantamine) are approved for mild to moderate Alzheimer disease, with donepezil also approved for severe disease. These agents provide modest cognitive and functional benefits but may cause gastrointestinal effects, bradycardia, and sleep disturbances. Memantine, an NMDA receptor antagonist, is approved for moderate to severe Alzheimer disease. Newer anti-amyloid monoclonal antibodies, such as aducanumab and lecanemab, target amyloid pathology and may modestly slow cognitive decline in early disease but require careful patient selection and monitoring due to safety concerns.
Patients with atypical presentations or complex management needs should be referred to specialists with expertise in dementia care. Ongoing caregiver education and support are critical, as they reduce caregiver burden, delay institutionalization, and improve patient outcomes. Prevention strategies emphasize physical activity, cardiovascular risk factor control, cognitive engagement, healthy diet patterns, and avoidance of tobacco and excessive alcohol use.
- Published on
KembaraXtra- Medicine – Allergic Rhinitis
Allergic rhinitis (AR) is an immunoglobulin E (IgE)–mediated hypersensitivity reaction to inhaled environmental allergens that leads to inflammation of the nasal mucosa. This inflammatory process is driven primarily by type 2 helper T (Th2) cells and results in the characteristic symptoms of sneezing, rhinorrhea, nasal pruritus, and nasal congestion. Allergic rhinitis may be seasonal, triggered by pollens, or perennial, caused by year-round allergens such as dust mites, molds, and animal dander.
Allergic rhinitis is highly prevalent, affecting 10% to 30% of the U.S. population and up to 40% of children, with substantial impact on quality of life, school performance, and workplace productivity. The mean age of onset is between 8 and 12 years, and approximately 80% of cases develop before age 20. Sensitization to aeroallergens is uncommon before the age of one year. Among patients presenting to primary care with nasal complaints, allergic rhinitis accounts for 30% to 60% of cases.
Clinically, allergic rhinitis is distinguished from nonallergic rhinitis by the presence of sneezing, nasal itching, and ocular pruritus. Both allergic and nonallergic rhinitis may cause nasal congestion, postnasal drip, and cough. Additional symptoms include fatigue, irritability, reduced sense of smell, and a sensation of ear fullness. Physical examination often reveals pale or violaceous, edematous nasal turbinates caused by venous engorgement, a feature that helps distinguish allergic rhinitis from viral rhinitis, which typically presents with erythematous mucosa. Other findings may include clear nasal discharge, posterior pharyngeal drainage, conjunctival injection, throat erythema, and occasionally nasal polyps.
The most common triggers of allergic rhinitis include pollens (trees in spring, grasses in summer, and weeds in fall), as well as dust mites, mold spores, and animal allergens. Nonallergic irritants such as smoke, strong odors, and pollution may exacerbate symptoms in both allergic and nonallergic rhinitis but do not represent the primary mechanism of disease.
Diagnosis is largely clinical, based on history and physical examination. Many patients do not require formal diagnostic testing if symptoms and triggers are typical. A seasonal pattern of symptoms can help identify the likely allergen source, whereas year-round symptoms suggest perennial allergens. Allergy testing, either by skin testing or serum-specific IgE testing, may be useful in patients with persistent symptoms despite standard therapy, those considering allergen immunotherapy, or when precise identification of allergens is necessary for avoidance strategies.
Management begins with nonpharmacologic measures, including allergen avoidance when feasible. Strategies include dust mite control with mattress and pillow encasements, removal of carpeting and allergen-collecting items from the bedroom, use of HEPA air filtration, and limiting exposure to pets in sensitized individuals. Nasal saline sprays or irrigations are effective adjunctive therapies that reduce nasal dryness, congestion, and allergen load.
For pharmacologic treatment, intranasal corticosteroids are the preferred first-line therapy for most patients with allergic rhinitis. These agents effectively reduce nasal inflammation and improve all major symptoms, although maximal benefit may take at least one week of regular use. In patients with moderate to severe disease, combination therapy with an intranasal corticosteroid and an intranasal antihistamine provides greater symptom control than either agent alone. Oral second-generation antihistamines may be helpful for persistent ocular symptoms but offer little additional benefit when combined with intranasal corticosteroids for nasal symptoms. Montelukast should be reserved for patients who cannot tolerate or do not respond to first-line therapies, and intranasal ipratropium may be considered when rhinorrhea is the predominant complaint.
For patients with persistent or severe symptoms despite optimal medical therapy, allergen immunotherapy may be considered. Both subcutaneous and sublingual immunotherapy can significantly reduce symptoms and medication use, with approximately 80% of patients reporting improvement. Immunotherapy is contraindicated in patients with uncontrolled asthma, a history of severe reactions to immunotherapy, and—specifically for sublingual therapy—those with eosinophilic esophagitis.
Overall, the prognosis of allergic rhinitis is excellent. Most patients achieve good symptom control with appropriate avoidance strategies and medical therapy. Referral to an allergist or otolaryngologist is appropriate for patients with moderate to severe disease, those unresponsive to treatment, or individuals interested in immunotherapy.

- Published on
KembaraXtra- Medicine – Alpha-1-Antitrypsin Deficiency
Alpha-1-antitrypsin deficiency (AATD) is an inherited genetic disorder caused by deficiency or dysfunction of the protease inhibitor alpha-1-antitrypsin (AAT). The condition most commonly results from homozygous carriage of the SERPINA1 Z allele (PI*ZZ genotype). This mutation leads to production of an abnormal AAT protein (Z-AAT) that accumulates within hepatocytes while circulating levels of functional AAT are markedly reduced. The resulting imbalance predisposes affected individuals to early-onset pulmonary emphysema, chronic obstructive pulmonary disease (COPD), and progressive liver disease, including cirrhosis and hepatocellular carcinoma.
Alpha-1-antitrypsin deficiency is underrecognized but accounts for approximately 1% to 5% of COPD cases in the United States, with an estimated prevalence of 1 in 2000 to 5000 individuals. The disorder is inherited in an autosomal codominant fashion. The normal M allele is present in about 95% of the population, while the deficient Z allele occurs in 1% to 2% and the S allele in 2% to 3%. Severe deficiency most often occurs in individuals with the ZZ genotype, while those with SZ or MZ genotypes may have partial deficiency, with increased risk of lung disease—particularly in smokers. Approximately 1 in 10 individuals of European descent carries at least one abnormal allele.
Clinical manifestations vary depending on genotype and environmental exposures. The lungs are most commonly affected, with patients developing early-onset, lower-lobe–predominant panlobular emphysema, often before age 45 and sometimes in the absence of smoking. Symptoms resemble typical COPD and include progressive dyspnea, chronic cough, and sputum production. Bronchiectasis may also be present. Smoking significantly accelerates lung damage and worsens prognosis. Liver involvement may present as neonatal cholestasis, chronic hepatitis, cirrhosis, or hepatocellular carcinoma in both children and adults. A rare dermatologic manifestation is panniculitis, characterized by painful, inflammatory subcutaneous nodules caused by unopposed proteolysis in the skin.
The pathophysiology of AATD involves loss of antiprotease protection in the lungs and toxic accumulation of mutant AAT protein in hepatocytes. In the lungs, reduced AAT levels fail to adequately inhibit neutrophil elastase, leading to destruction of alveolar elastin and development of emphysema. In the liver, disease results not from deficiency but from intracellular retention of misfolded Z-AAT, which triggers hepatocellular injury, inflammation, and fibrosis. Not all individuals with AAT deficiency develop clinical disease, but smoking is the strongest modifiable risk factor for lung involvement.
Diagnosis should be suspected in patients with early-onset COPD, emphysema without typical risk factors, or basilar-predominant emphysema on imaging. Unexplained liver disease, panniculitis, bronchiectasis, poorly responsive asthma, or c-ANCA–associated vasculitis should also prompt evaluation. Initial testing includes measurement of serum alpha-1-antitrypsin levels, followed by genotyping to identify specific alleles. Pulmonary function testing typically shows airflow obstruction consistent with COPD. Imaging with chest radiography or high-resolution computed tomography often demonstrates lower-lobe–predominant emphysema and may reveal bronchiectasis.
Management begins with nonpharmacologic interventions, most importantly complete avoidance of smoking, which dramatically reduces the risk and progression of lung disease. Patients should also avoid occupational and environmental exposures that damage the lungs and receive routine vaccinations, including influenza, pneumococcal, pertussis, and SARS-CoV-2 vaccines. Acute COPD exacerbations are treated similarly to those in patients without AATD.
For chronic management, the therapeutic goal is to raise serum AAT levels above the protective threshold of approximately 50 mg/dL. Currently, the only FDA-approved disease-specific therapy is intravenous alpha-1-antitrypsin augmentation therapy, which uses pooled human AAT to slow progression of emphysema. This treatment requires lifelong administration, is costly, and does not reverse established lung damage nor improve liver disease. Patients with end-stage lung or liver disease may be candidates for organ transplantation, which can be life-saving and curative for the affected organ.
Prognosis depends on genotype, degree of deficiency, and environmental factors. Among patients with severe AATD, respiratory failure is the leading cause of death, followed by cirrhosis and liver-related complications. Early diagnosis, smoking cessation, and appropriate specialty care significantly improve outcomes. Referral to clinicians experienced in managing AATD is recommended, including pulmonology, hepatology, and transplant centers when indicated.
Alpha-1-antitrypsin deficiency remains substantially underdiagnosed, and expert guidelines recommend testing all patients with COPD, emphysema, irreversible asthma, unexplained liver disease, bronchiectasis, or panniculitis, as well as first-degree relatives of affected individuals. Early recognition allows for risk reduction, appropriate monitoring, and timely intervention to prevent irreversible organ damage.
- Published on
KembaraXtra- Medicine – Alopecia
Alopecia refers to involuntary hair loss, most commonly affecting the scalp but potentially involving any hair-bearing area of the body. Alopecia is broadly classified into nonscarring (noncicatricial) and scarring (cicatricial) forms. Nonscarring alopecia is characterized by hair loss without permanent follicular destruction, scarring, or skin atrophy, whereas scarring alopecia results in irreversible hair loss due to follicular damage accompanied by inflammation, fibrosis, and loss of follicular openings.
Among nonscarring alopecias, alopecia areata is an autoimmune condition causing patchy hair loss on the scalp, eyebrows, eyelashes, or beard area, and may progress to alopecia totalis (complete scalp hair loss) or alopecia universalis (complete loss of body hair). Androgenetic alopecia (AGA) is the most common form of hair loss and results from genetically determined follicular sensitivity to androgens. In men, it presents as bitemporal recession, frontal thinning, and vertex balding (male pattern hair loss), while in women it manifests as diffuse central scalp thinning with preservation of the frontal hairline (female pattern hair loss). Women may initially notice increased shedding and reduced ponytail volume years before visible thinning develops.
The epidemiology of alopecia varies by etiology. Alopecia areata affects approximately 1% of the U.S. population by age 50 and occurs equally in both sexes. Androgenetic alopecia affects up to 50% of Caucasian men by age 50, with lower prevalence and later onset in Asian and African-American men. By age 70, approximately 40% of women are affected, particularly after menopause. Genetic factors play a significant role, especially in androgenetic alopecia, which is polygenic with variable penetrance, while certain scarring alopecias are more common in individuals with coarse or tightly curled hair.
The causes of alopecia are diverse and include autoimmune disorders, hormonal changes, infections, medications, nutritional deficiencies, physical or psychological stress, and trauma. Nonscarring alopecia may result from altered hair cycling (as in telogen effluvium or anagen effluvium), follicular miniaturization (as in androgenetic alopecia), hair shaft damage (from traction, trichotillomania, or cosmetic overprocessing), or immune-mediated follicular attack (as in alopecia areata). Scarring alopecia may be caused by inflammatory or autoimmune diseases such as lichen planopilaris or discoid lupus erythematosus, infections like severe tinea capitis with kerion formation, neoplasms, or congenital conditions.
Clinical evaluation begins with a thorough history, focusing on the onset, duration, and pattern of hair loss; recent stressors or illnesses; medication use; nutritional status; hair care practices; family history; and associated systemic or dermatologic symptoms. Physical examination assesses the distribution and extent of hair loss, presence of inflammation or scarring, hair shaft abnormalities, and associated findings such as virilization in women or nail changes in alopecia areata. Characteristic findings include exclamation-point hairs in alopecia areata, broken hairs of varying lengths in traumatic alopecia, and shiny atrophic skin with loss of follicular openings in scarring alopecia.
Diagnostic evaluation may include a hair pull test, where removal of six or more hairs from a gentle pull suggests active shedding, most commonly seen in telogen effluvium. Scalp biopsy is essential when scarring alopecia is suspected and should be performed with both vertical and horizontal sections for optimal histopathologic assessment. A trichogram may help quantify the proportion of anagen versus telogen hairs. Laboratory testing is guided by clinical suspicion and may include complete blood count, iron studies, thyroid function tests, autoimmune markers, and hormonal evaluation in women with signs of hyperandrogenism.
The differential diagnosis of nonscarring alopecia includes telogen effluvium, characterized by diffuse shedding following physiologic or psychological stress; androgenetic alopecia, with its characteristic patterned thinning; alopecia areata, presenting with well-demarcated patches of hair loss and autoimmune features; tinea capitis, especially in children; and traumatic alopecia, including traction alopecia and trichotillomania. Scarring alopecias include conditions such as lichen planopilaris, discoid lupus erythematosus, and inflammatory tinea capitis with kerion formation, all of which may lead to permanent hair loss if untreated.
Treatment depends on the underlying cause. Telogen effluvium is typically self-limited, with hair regrowth occurring within months once the inciting factor is removed. Androgenetic alopecia is managed with topical minoxidil in both sexes and oral finasteride in men; discontinuation of therapy results in recurrence of hair loss. Additional options include antiandrogens in selected women, hair transplantation, cosmetic camouflage, and low-level light therapy. Alopecia areata may resolve spontaneously, but treatments include topical, intralesional, or systemic corticosteroids, topical immunotherapy, and newer agents such as oral JAK inhibitors, which are now approved for severe disease in adults. Tinea capitis requires systemic antifungal therapy, and inflammatory kerion may necessitate adjunctive corticosteroids. Traction alopecia improves with elimination of the offending hairstyle or practice, while scarring alopecias focus on early inflammation control to prevent further permanent loss.
Patients with suspected scarring alopecia, treatment-resistant disease, or diagnostic uncertainty should be referred to dermatology. Education and counseling are essential, as hair loss can have a profound psychological impact. For patients with alopecia areata, support organizations and patient education resources can provide valuable guidance and reassurance.

- Published on
KembaraXtra-Medicine- Anorectal Stricture
Basic Information and Definition
An anorectal stricture, also referred to as anal stenosis, is an abnormal narrowing of the anal canal. Strictures are classified according to both severity and location. Severity ranges from mild, in which a well-lubricated finger or medium Hill-Ferguson retractor can pass easily, to moderate, where passage is possible only with forceful dilation, and severe, where no passage is possible. Location is described as low (at least 0.5 cm distal to the dentate line), middle (within 0.5 cm on either side of the dentate line), or high (at least 0.5 cm proximal to the dentate line).
Synonyms and Coding
Anorectal stricture is also known as anal stenosis or Küss disease. The ICD-10-CM code used is K62.4, which denotes stenosis of the anus and rectum.
Epidemiology and Risk Factors
Hemorrhoid surgery is the most common overall cause of anorectal stricture, occurring in approximately 5% to 10% of patients after radical hemorrhoidectomy, particularly for grade III and IV hemorrhoidal disease. The risk of developing a stricture is directly related to the extent of anoderm resection. Other important risk factors include prior anorectal surgery, inflammatory bowel disease, previous pelvic radiation, and chronic hemorrhoidal disease.
Physical Findings and Clinical Presentation
Pain with defecation is the most common presenting symptom. Patients may also report rectal bleeding, narrowing of stools, severe constipation, outlet obstruction, fecal incontinence, tenesmus, or urgency. A careful history should assess prior anorectal surgery, Crohn disease, radiation exposure, or perianal trauma. On physical examination, visual inspection may reveal skin tags or chronic fissures. Digital rectal examination typically demonstrates a narrowed anal canal with difficulty passing a lubricated finger. In severe cases, examination under anesthesia may be required to adequately assess the stricture.
Etiology
Anorectal strictures are broadly divided into congenital, primary, and secondary causes. Congenital strictures are pediatric conditions related to abnormal embryologic development, including anal atresia and imperforate anus. Primary strictures usually occur in elderly patients due to fibrous involution of perianal tissues. Secondary strictures are the most common and are typically iatrogenic, most often following hemorrhoidectomy or other anorectal surgeries such as low anterior resection, ileal pouch–anal anastomosis, anopexy, or excision of perianal skin lesions. Other secondary causes include fibrosis of the anoderm or distal rectal mucosa, anal canal muscle hypertrophy or spasm, neoplastic conditions (such as Bowen disease, Paget disease, anal squamous cell carcinoma, rectal adenocarcinoma, or condyloma acuminata), inflammatory diseases (including anal fissure, Crohn disease, tuberculosis, actinomycosis, lymphogranuloma venereum, and chronic suppuration), traumatic causes (radiation therapy, perineal burns, hot water enemas, ibuprofen suppositories, and chronic laxative abuse), and sexually transmitted infections.
Diagnosis and Differential Diagnosis
Anorectal stricture is primarily a clinical diagnosis. The differential diagnosis includes anorectal or rectal neoplasm, inflammatory bowel disease, and traumatic injury. Any suspicious lesions identified during examination should be biopsied to exclude malignancy or inflammatory pathology.
Workup
Although the diagnosis is clinical, evaluation should ensure that secondary causes such as neoplasm or inflammatory disease are excluded. Regardless of etiology, hydration, fiber supplementation, and stool softeners should be initiated in all patients to reduce symptoms and prevent further injury.
Treatment
Management depends on severity and underlying cause. Nonpharmacologic therapy includes manual dilation using digital techniques or commercial dilators, which may be initiated in the clinic or operating room and continued on an outpatient basis for mild to moderate strictures. Surgical options include resection of neoplasm, stricturoplasty, or anoplasty. While multiple operative techniques have been described, there is no consensus on a single superior approach; all aim to replace scar tissue with healthy, pliable anoderm within the anal canal.
Chronic Management
For mild disease, conservative measures such as stool softeners, fiber supplementation, adequate hydration, and dietary modification may be sufficient to control symptoms without the need for procedural intervention.
Prevention and Complementary Considerations
The most effective management of postsurgical anorectal stricture is prevention. Proper surgical technique during anorectal procedures, including preservation of anoderm and, in some cases, internal sphincterotomy, can significantly reduce the risk of developing stenosis.
Disposition and Referral
Mild strictures can usually be managed on an outpatient basis. Moderate to severe strictures require evaluation by a colorectal surgeon. Referral is also indicated when there is concern for neoplasm, failure of conservative management, or inability of the clinician to pass a lubricated finger into the rectum during examination.
Basic Information and Definition
An anorectal stricture, also referred to as anal stenosis, is an abnormal narrowing of the anal canal. Strictures are classified according to both severity and location. Severity ranges from mild, in which a well-lubricated finger or medium Hill-Ferguson retractor can pass easily, to moderate, where passage is possible only with forceful dilation, and severe, where no passage is possible. Location is described as low (at least 0.5 cm distal to the dentate line), middle (within 0.5 cm on either side of the dentate line), or high (at least 0.5 cm proximal to the dentate line).
Synonyms and Coding
Anorectal stricture is also known as anal stenosis or Küss disease. The ICD-10-CM code used is K62.4, which denotes stenosis of the anus and rectum.
Epidemiology and Risk Factors
Hemorrhoid surgery is the most common overall cause of anorectal stricture, occurring in approximately 5% to 10% of patients after radical hemorrhoidectomy, particularly for grade III and IV hemorrhoidal disease. The risk of developing a stricture is directly related to the extent of anoderm resection. Other important risk factors include prior anorectal surgery, inflammatory bowel disease, previous pelvic radiation, and chronic hemorrhoidal disease.
Physical Findings and Clinical Presentation
Pain with defecation is the most common presenting symptom. Patients may also report rectal bleeding, narrowing of stools, severe constipation, outlet obstruction, fecal incontinence, tenesmus, or urgency. A careful history should assess prior anorectal surgery, Crohn disease, radiation exposure, or perianal trauma. On physical examination, visual inspection may reveal skin tags or chronic fissures. Digital rectal examination typically demonstrates a narrowed anal canal with difficulty passing a lubricated finger. In severe cases, examination under anesthesia may be required to adequately assess the stricture.
Etiology
Anorectal strictures are broadly divided into congenital, primary, and secondary causes. Congenital strictures are pediatric conditions related to abnormal embryologic development, including anal atresia and imperforate anus. Primary strictures usually occur in elderly patients due to fibrous involution of perianal tissues. Secondary strictures are the most common and are typically iatrogenic, most often following hemorrhoidectomy or other anorectal surgeries such as low anterior resection, ileal pouch–anal anastomosis, anopexy, or excision of perianal skin lesions. Other secondary causes include fibrosis of the anoderm or distal rectal mucosa, anal canal muscle hypertrophy or spasm, neoplastic conditions (such as Bowen disease, Paget disease, anal squamous cell carcinoma, rectal adenocarcinoma, or condyloma acuminata), inflammatory diseases (including anal fissure, Crohn disease, tuberculosis, actinomycosis, lymphogranuloma venereum, and chronic suppuration), traumatic causes (radiation therapy, perineal burns, hot water enemas, ibuprofen suppositories, and chronic laxative abuse), and sexually transmitted infections.
Diagnosis and Differential Diagnosis
Anorectal stricture is primarily a clinical diagnosis. The differential diagnosis includes anorectal or rectal neoplasm, inflammatory bowel disease, and traumatic injury. Any suspicious lesions identified during examination should be biopsied to exclude malignancy or inflammatory pathology.
Workup
Although the diagnosis is clinical, evaluation should ensure that secondary causes such as neoplasm or inflammatory disease are excluded. Regardless of etiology, hydration, fiber supplementation, and stool softeners should be initiated in all patients to reduce symptoms and prevent further injury.
Treatment
Management depends on severity and underlying cause. Nonpharmacologic therapy includes manual dilation using digital techniques or commercial dilators, which may be initiated in the clinic or operating room and continued on an outpatient basis for mild to moderate strictures. Surgical options include resection of neoplasm, stricturoplasty, or anoplasty. While multiple operative techniques have been described, there is no consensus on a single superior approach; all aim to replace scar tissue with healthy, pliable anoderm within the anal canal.
Chronic Management
For mild disease, conservative measures such as stool softeners, fiber supplementation, adequate hydration, and dietary modification may be sufficient to control symptoms without the need for procedural intervention.
Prevention and Complementary Considerations
The most effective management of postsurgical anorectal stricture is prevention. Proper surgical technique during anorectal procedures, including preservation of anoderm and, in some cases, internal sphincterotomy, can significantly reduce the risk of developing stenosis.
Disposition and Referral
Mild strictures can usually be managed on an outpatient basis. Moderate to severe strictures require evaluation by a colorectal surgeon. Referral is also indicated when there is concern for neoplasm, failure of conservative management, or inability of the clinician to pass a lubricated finger into the rectum during examination.


