- Published on
KembaraXtra-Medicine – ANCA-Associated Vasculitis
Basic Information Anti-neutrophil cytoplasmic antibody (ANCA)–associated vasculitis (AAV) is a group of small- to medium-vessel systemic vasculitides that includes granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA). These conditions share overlapping clinical manifestations and treatment strategies but differ in pathology and organ involvement. They are characterized by necrotizing, pauci-immune small-vessel vasculitis, with granulomatous inflammation present in GPA but absent in MPA. Most patients present with renal and pulmonary involvement, although the skin, nervous system, and gastrointestinal tract may also be affected. A limited form of GPA may be confined to the upper respiratory tract and can often be managed less aggressively.
Synonyms ANCA-associated vasculitis, ANCA-positive vasculitis, granulomatosis with polyangiitis (GPA), Wegener granulomatosis, microscopic polyangiitis (MPA), eosinophilic granulomatosis with polyangiitis (EGPA).
ICD-10-CM Codes I77.6 arteritis unspecified; M31.3 granulomatosis with polyangiitis; M31.7 microscopic polyangiitis; M30.1 eosinophilic granulomatosis with polyangiitis.
Epidemiology and Demographics The incidence is approximately 6–12 cases per million persons per year in Western Europe and Japan, with global variation. Prevalence ranges from 94 to 160 per million. There is a slight male predominance, and incidence increases with age, peaking between 60 and 70 years.
Risk Factors and Genetics Risk factors include infections such as Staphylococcus aureus, silica and hydrocarbon exposure, cigarette smoking, pesticides, and medications including hydralazine, minocycline, propylthiouracil, levamisole-adulterated cocaine, and allopurinol. Familial disease is rare. GPA is associated with SNPs in HLA-DP, PRTN3, and SERPINA1, whereas MPA is associated with HLA-DQ polymorphisms.
Physical Findings and Clinical Presentation Patients commonly have constitutional symptoms such as fever, fatigue, weight loss, myalgia, and arthralgia. Upper respiratory tract disease is prominent in GPA and includes chronic sinusitis, otitis media, mastoiditis, nasal crusting and epistaxis, septal perforation, subglottic or tracheal stenosis, and saddle-nose deformity. Ocular involvement includes conjunctivitis, episcleritis, scleritis, uveitis, optic neuropathy, and retinal vasculitis. Ear findings include sensorineural or conductive hearing loss and otorrhea. Oral findings may include chronic ulcerative lesions and “mulberry” gingivitis. Pulmonary disease presents with dyspnea, hemoptysis, pleuritic chest pain, nodules, cavitary lesions, infiltrates, effusions, or diffuse alveolar hemorrhage. Gastrointestinal involvement, more common in MPA, includes ischemic abdominal pain, bleeding, ulceration, or perforation. Renal disease is common in GPA and MPA and manifests as hypertension, edema, reduced urine output, rapidly progressive glomerulonephritis, microscopic hematuria, red cell casts, and proteinuria. Neurologic involvement includes mononeuritis multiplex, peripheral neuropathy, pachymeningitis, headaches, and rarely central nervous system disease. Cutaneous findings include palpable purpura, necrotic ulcers, nodules, urticaria, livedo reticularis, and digital gangrene.
Etiology The etiology is immune mediated and multifactorial. ANCAs directed against proteinase 3 (PR3) or myeloperoxidase (MPO) activate neutrophils, causing endothelial injury and tissue necrosis. Genetic susceptibility and environmental triggers contribute to disease development.
Diagnosis and Differential Diagnosis
Diagnosis is based on compatible clinical features supported by laboratory testing, imaging, and histopathology when feasible. Differential diagnoses include other systemic vasculitides, granulomatous lung diseases, anti–glomerular basement membrane disease, infections, malignancy, and drug- or cocaine-induced vasculitis.
Workup Evaluation
includes detailed history and physical examination, chest imaging, renal assessment, and targeted organ evaluation. ANCA serology typically shows PR3-ANCA in GPA and MPO-ANCA in MPA, though overlap exists and up to 15% of patients may be ANCA negative. Laboratory findings include anemia, leukocytosis, thrombocytosis, elevated inflammatory markers, impaired renal function, hematuria, and proteinuria. Tissue biopsy of affected organs, particularly kidney or lung, is recommended when safe to confirm diagnosis.
Treatment Management depends on disease severity and organ involvement. Induction therapy for moderate to severe disease includes systemic glucocorticoids combined with rituximab or cyclophosphamide. Reduced-dose steroid regimens may be used. Plasma exchange may be considered in select severe cases, though overall benefit is limited. Mild or limited disease may be treated with methotrexate-based regimens. Maintenance therapy commonly includes rituximab, azathioprine, methotrexate, or mycophenolate mofetil and is generally continued for 18–24 months. EGPA may additionally be treated with anti–interleukin-5 therapy such as mepolizumab. Prophylaxis against Pneumocystis jirovecii pneumonia is recommended during intensive immunosuppression.
Disposition and Referral
With prompt treatment, remission is achieved in most patients, though relapse is common. Renal failure and pulmonary involvement are major determinants of prognosis. Referral to rheumatology is essential, with multidisciplinary involvement from nephrology, pulmonology, ENT, dermatology, and neurology as indicated.
Pearls and Considerations
The goal of therapy is rapid induction of durable remission followed by maintenance treatment. Without treatment, mortality is high, and therapy is complicated by infection risk and progressive organ damage. Support and education resources are available through organizations such as the Vasculitis Foundation.
Basic Information Anti-neutrophil cytoplasmic antibody (ANCA)–associated vasculitis (AAV) is a group of small- to medium-vessel systemic vasculitides that includes granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA). These conditions share overlapping clinical manifestations and treatment strategies but differ in pathology and organ involvement. They are characterized by necrotizing, pauci-immune small-vessel vasculitis, with granulomatous inflammation present in GPA but absent in MPA. Most patients present with renal and pulmonary involvement, although the skin, nervous system, and gastrointestinal tract may also be affected. A limited form of GPA may be confined to the upper respiratory tract and can often be managed less aggressively.
Synonyms ANCA-associated vasculitis, ANCA-positive vasculitis, granulomatosis with polyangiitis (GPA), Wegener granulomatosis, microscopic polyangiitis (MPA), eosinophilic granulomatosis with polyangiitis (EGPA).
ICD-10-CM Codes I77.6 arteritis unspecified; M31.3 granulomatosis with polyangiitis; M31.7 microscopic polyangiitis; M30.1 eosinophilic granulomatosis with polyangiitis.
Epidemiology and Demographics The incidence is approximately 6–12 cases per million persons per year in Western Europe and Japan, with global variation. Prevalence ranges from 94 to 160 per million. There is a slight male predominance, and incidence increases with age, peaking between 60 and 70 years.
Risk Factors and Genetics Risk factors include infections such as Staphylococcus aureus, silica and hydrocarbon exposure, cigarette smoking, pesticides, and medications including hydralazine, minocycline, propylthiouracil, levamisole-adulterated cocaine, and allopurinol. Familial disease is rare. GPA is associated with SNPs in HLA-DP, PRTN3, and SERPINA1, whereas MPA is associated with HLA-DQ polymorphisms.
Physical Findings and Clinical Presentation Patients commonly have constitutional symptoms such as fever, fatigue, weight loss, myalgia, and arthralgia. Upper respiratory tract disease is prominent in GPA and includes chronic sinusitis, otitis media, mastoiditis, nasal crusting and epistaxis, septal perforation, subglottic or tracheal stenosis, and saddle-nose deformity. Ocular involvement includes conjunctivitis, episcleritis, scleritis, uveitis, optic neuropathy, and retinal vasculitis. Ear findings include sensorineural or conductive hearing loss and otorrhea. Oral findings may include chronic ulcerative lesions and “mulberry” gingivitis. Pulmonary disease presents with dyspnea, hemoptysis, pleuritic chest pain, nodules, cavitary lesions, infiltrates, effusions, or diffuse alveolar hemorrhage. Gastrointestinal involvement, more common in MPA, includes ischemic abdominal pain, bleeding, ulceration, or perforation. Renal disease is common in GPA and MPA and manifests as hypertension, edema, reduced urine output, rapidly progressive glomerulonephritis, microscopic hematuria, red cell casts, and proteinuria. Neurologic involvement includes mononeuritis multiplex, peripheral neuropathy, pachymeningitis, headaches, and rarely central nervous system disease. Cutaneous findings include palpable purpura, necrotic ulcers, nodules, urticaria, livedo reticularis, and digital gangrene.
Etiology The etiology is immune mediated and multifactorial. ANCAs directed against proteinase 3 (PR3) or myeloperoxidase (MPO) activate neutrophils, causing endothelial injury and tissue necrosis. Genetic susceptibility and environmental triggers contribute to disease development.
Diagnosis and Differential Diagnosis
Diagnosis is based on compatible clinical features supported by laboratory testing, imaging, and histopathology when feasible. Differential diagnoses include other systemic vasculitides, granulomatous lung diseases, anti–glomerular basement membrane disease, infections, malignancy, and drug- or cocaine-induced vasculitis.
Workup Evaluation
includes detailed history and physical examination, chest imaging, renal assessment, and targeted organ evaluation. ANCA serology typically shows PR3-ANCA in GPA and MPO-ANCA in MPA, though overlap exists and up to 15% of patients may be ANCA negative. Laboratory findings include anemia, leukocytosis, thrombocytosis, elevated inflammatory markers, impaired renal function, hematuria, and proteinuria. Tissue biopsy of affected organs, particularly kidney or lung, is recommended when safe to confirm diagnosis.
Treatment Management depends on disease severity and organ involvement. Induction therapy for moderate to severe disease includes systemic glucocorticoids combined with rituximab or cyclophosphamide. Reduced-dose steroid regimens may be used. Plasma exchange may be considered in select severe cases, though overall benefit is limited. Mild or limited disease may be treated with methotrexate-based regimens. Maintenance therapy commonly includes rituximab, azathioprine, methotrexate, or mycophenolate mofetil and is generally continued for 18–24 months. EGPA may additionally be treated with anti–interleukin-5 therapy such as mepolizumab. Prophylaxis against Pneumocystis jirovecii pneumonia is recommended during intensive immunosuppression.
Disposition and Referral
With prompt treatment, remission is achieved in most patients, though relapse is common. Renal failure and pulmonary involvement are major determinants of prognosis. Referral to rheumatology is essential, with multidisciplinary involvement from nephrology, pulmonology, ENT, dermatology, and neurology as indicated.
Pearls and Considerations
The goal of therapy is rapid induction of durable remission followed by maintenance treatment. Without treatment, mortality is high, and therapy is complicated by infection risk and progressive organ damage. Support and education resources are available through organizations such as the Vasculitis Foundation.

- Published on
KembaraXtra-Medicine – Anaphylaxis
Anaphylaxis is a severe, rapid-onset, life-threatening allergic reaction. It occurs when IgE-mediated and non–IgE-mediated systemic mast cell degranulation leads to release of inflammatory mediators, producing respiratory, cardiovascular, gastrointestinal, and/or mucocutaneous symptoms. A common synonym is anaphylactic reaction. Relevant ICD-10CM codes include T78.2 (anaphylactic shock, unspecified, initial encounter), T78.00XA (anaphylactic reaction due to unspecified food, initial encounter), T80.52XA (anaphylactic reaction due to vaccination, initial encounter), and T63.94XA (toxic effect of contact with unspecified venomous animal, undetermined, initial encounter).
In the United States, the incidence is estimated at 50 to 2000 episodes per 100,000 persons, with a lifetime prevalence of 1.6% to 5.1%. Emergency department visits for anaphylaxis increased by 101% from 2005 to 2014, yet fatalities have remained stable at about 0.7 per million adults per year. In adults, medications and insect stings are leading triggers, while in children and adolescents, foods and insect stings are the leading triggers.
Risk factors depend on the trigger and severity. Atopy increases risk for anaphylaxis triggered by food, exercise, and latex. Risk factors for severe anaphylaxis include cardiovascular disease, asthma, older age, mast cell disorder, beta-blocker use, and ACE inhibitor use.
Clinical presentation commonly includes involvement across multiple systems. Mucocutaneous findings include urticaria, pruritus, flushing, and angioedema. Respiratory symptoms include dyspnea, cough, wheezing, hypoxia, stridor, and rhinitis. Cardiovascular features include hypotension, tachycardia, weakness, dizziness, syncope, malaise, and vascular collapse. Gastrointestinal symptoms include nausea, vomiting, diarrhea, dysphagia, and abdominal pain.
Anaphylaxis results from a sudden systemic release of histamine and other mediators from basophils and mast cells via IgE-dependent and non–IgE-dependent mechanisms, and almost any substance can trigger it. In acute settings, the cause is often not identified, accounting for 30% to 60% of cases. Common triggers include foods and additives (notably peanuts, tree nuts, eggs, shellfish, fish, cow’s milk, fruits, soy). A notable food-related mechanism is alpha-gal syndrome, where a lone star tick bite can cause IgE sensitization to alpha-1,3-galactose, leading to delayed anaphylaxis after eating mammalian (red meat) products. Medication triggers include antibiotics (especially penicillins and sulfa agents), insulin, allergen extracts, opiates, vaccines, NSAIDs, contrast media, streptokinase, immunomodulators, and IV iron. Environmental exposures include bee/wasp stings, snake venom, and fire ant venom. Other triggers include blood products (plasma, immunoglobulin, cryoprecipitate, whole blood), latex, and exercise. Mechanistically, immune (often IgE-mediated) triggers include foods, venoms, many medications, latex, seminal fluid, and sometimes radiocontrast; IgE-independent immune triggers include radiocontrast (in most cases), dextrans, some monoclonal antibodies (e.g., rituximab), and some medications; and nonimmunologic triggers include certain drugs such as opioids and protamine. “Idiopathic” anaphylaxis refers to cases where known causes (including mastocytosis) have been excluded. Non–IgE mechanisms may involve complement activation, kinin production, potentiation, or direct mediator release, and more than one mechanism can occur together (for example, exercise-induced reactions following food). ACE inhibitors are a notable cause of unexplained angioedema, occurring in up to 1 in 200 users and may occur at any time after starting the medication.
The differential diagnosis is broad and includes allergic reactions without anaphylaxis, other causes of shock such as sepsis or pulmonary embolism, endocrine conditions such as carcinoid, adrenal crisis, paradoxical pheochromocytoma, systemic mastocytosis, serum sickness, severe asthma (distinguished by abrupt onset in anaphylaxis versus progressive worsening), scombroid poisoning, localized angioedema, acute urticaria, vasovagal/presyncopal syndromes, airway foreign body, vocal cord dysfunction, and anxiety-related symptoms such as globus hystericus.
Workup is mainly aimed at excluding mimics, but treatment should never be delayed because anaphylaxis can be rapidly fatal. Diagnosis is primarily clinical. Laboratory testing is generally not helpful in the emergency setting, though serum/urine histamine and serum tryptase (drawn 30 minutes to 2 hours after symptom onset and compared with baseline) may support the diagnosis; importantly, normal levels do not exclude anaphylaxis and these tests may not be readily available. Imaging is usually not helpful; chest radiography is considered when evaluating foreign body aspiration or pulmonary pathology in acute respiratory compromise. An ECG should be considered in patients with sudden loss of consciousness, chest pain, dyspnea, and in elderly patients; ECG typically shows sinus tachycardia.
Clinical criteria make anaphylaxis highly likely when any one of the following is met: (1) an acute illness with skin/mucosal involvement plus either respiratory compromise or reduced blood pressure/end-organ dysfunction symptoms; (2) rapid onset of two or more after exposure to a likely allergen--skin/mucosal involvement, respiratory compromise, reduced blood pressure, or persistent GI symptoms such as crampy abdominal pain/vomiting; or (3) reduced blood pressure after exposure to a known allergen--age-specific hypotension or >30% systolic BP decrease in infants/children, or systolic BP <90 mm hg or>30% decrease from baseline in adults.
Immediate nonpharmacologic treatment begins with removing the trigger, rapidly establishing and protecting the airway, providing supplemental oxygen if needed, obtaining IV access, giving IV fluids (normal saline), and placing the patient on cardiac monitoring.
The cornerstone medication is intramuscular epinephrine. Adults and children >30 kg should receive 0.3 to 0.5 mg IM epinephrine (1:1000 concentration) in the anterolateral thigh; anyone >50 kg should receive 0.5 mg IM. Children <30 kg should receive 0.01 mg />g IM epinephrine (1:1000 concentration). IM administration is preferred because it produces a faster and more reliable rise to effective plasma levels, and the dose can be repeated within minutes if symptoms do not improve. Adjunct therapies include H1 and H2 antihistamines such as diphenhydramine and famotidine to improve itching and erythema, but they do not reverse airway obstruction, respiratory compromise, or hypotension, and their onset is delayed 1–2 hours. Corticosteroids do not help acutely because of slow onset and have not been shown to prevent prolonged or biphasic anaphylaxis, but they are commonly used as secondary treatment. Aerosolized beta-agonists help treat bronchospasm. If hypotension or cardiovascular collapse persists despite fluids and IM epinephrine, IV epinephrine (1:10,000 concentration) is used as vasopressor therapy, and small IV boluses may be used in the peri-arrest state. Patients on beta-blockers may respond poorly to initial treatment; in these cases, IV glucagon should be considered.
Disposition depends on severity and risk of recurrence. Patients with mild episodes should be observed for 1 hour after symptom resolution. Patients with severe symptoms—including cyanosis, pulse oximetry <92%, systolic bp <90 mm hg, confusion, loss of consciousness, or incontinence—those needing more than one epinephrine dose, those with a wide pulse pressure, unknown trigger, cutaneous signs />ymptoms, or a medication trigger in children should be observed for at least 6 hours to watch for biphasic anaphylaxis.
For discharge planning and chronic management, patients who are stable and remain asymptomatic should be prescribed two epinephrine autoinjectors, with clear instructions on when and how to use them. Referral to an allergist is helpful when the trigger is unclear in the emergency setting. Patient education is essential: a prior history of anaphylaxis or known triggers is the most reliable way to identify risk. Patients should be instructed to carry an epinephrine device at all times; school-aged children should have an additional autoinjector available at school with appropriate staff. Patients should also be advised to wear or carry MedicAlert identification listing substances that have triggered anaphylaxis. People with prior contrast reactions should avoid radiologic contrast when possible; however, pretreatment regimens with methylprednisolone or diphenhydramine may be used when contrast is unavoidable. For insect-sting anaphylaxis, venom immunotherapy initiated after the event is effective and is recommended for up to 5 years after the incident.

- Published on
Emergency And Acute Medicine – Inborn Errors Of Metabolism
Basics
Description Inborn errors of metabolism are inherited defects in metabolic pathways that lead to abnormal accumulation or deficiency of metabolites. More than 400 disorders are described, most presenting in infancy or childhood with variable and often nonspecific findings. Incidence ranges from 1:10,000 to 1:200,000 births. Major categories include amino acid disorders, urea cycle defects, organic acidemias, fatty acid oxidation defects, mitochondrial disorders, carbohydrate disorders, lysosomal storage diseases, peroxisomal disorders, and disorders of protein glycosylation. Pathophysiology relates directly to enzymatic or transport defects within metabolic pathways.
Etiology Genetic deficiency of enzymes or membrane transport systems involved in intermediary metabolism.
Diagnosis
Signs and symptoms Presentation may be acute and catastrophic or chronic and indolent. Neonates may initially be asymptomatic or present with hypothermia (mitochondrial defects), hypotonia or hypertonia (peroxisomal disorders), apnea (urea cycle defects, organic acidemias), seizures, vomiting, poor feeding, jaundice, hypoglycemia, dysmorphic features, or coma. Older untreated children may present with failure to thrive, dehydration, recurrent vomiting or diarrhea, food intolerance, lethargy, ataxia, seizures, or developmental delay.
History Review newborn screening results, dietary history, family history, consanguinity, and intercurrent illness.
Physical exam May reveal abnormal odor, altered mental status, tachypnea, dysmorphic facies, cataracts, cardiomyopathy, hepatosplenomegaly, dermatitis, or jaundice.
Essential workup Consider in any child with unexplained neurologic deterioration, persistent vomiting, metabolic acidosis, hypoglycemia, shock unresponsive to resuscitation, or failure to thrive.
Diagnosis tests and interpretation
Lab Bedside glucose, electrolytes, BUN/creatinine, CBC, calcium, liver function tests, bilirubin, coagulation studies, blood gas, lactate, pyruvate, uric acid, urinalysis, ammonia, serum amino acids, urine organic acids, and cultures as indicated.
Imaging CT head for altered mental status; chest radiograph as indicated.
Procedures Lumbar puncture when infection is a concern.
Differential diagnosis Sepsis, meningitis, encephalitis, dehydration, toxic ingestion, nonaccidental trauma, Reye syndrome, hepatic encephalopathy, endocrine disorders, renal failure, renal tubular acidosis, CNS mass lesions.
Treatment
Prehospital Prioritize ABCs and bedside glucose. Start IV glucose immediately; do not delay for fluids unless in shock. Avoid lactated Ringer solution. Keep patient NPO.
Initial stabilization Administer glucose (after Accu-Chek and thiamine when appropriate) and naloxone for altered mental status.
Emergency department management Secure airway, breathing, and circulation. Use normal saline for fluid boluses and avoid hypotonic fluids and lactated Ringer. Initiate IV glucose at 8–10 mg/kg/min (D10 at 1.5× maintenance) to prevent catabolism. Treat severe hypoglycemia with D25 bolus. Correct dehydration and acid–base abnormalities; consider bicarbonate only if pH <7.0. initiate dialysis for refractory severe acidosis or hyperammonemia. stop all oral intake initially. treat hyperammonemia urgently with ammonia-scavenging agents (arginine, sodium benzoate, phenylacetate, phenylbutyrate) in consultation a metabolic specialist. identify and precipitating infections.< />pan>
Medication D25 2–4 mL/kg IV for hypoglycemia, sodium bicarbonate 1–2 mEq/kg IV for severe acidosis, disease-specific therapies such as levocarnitine or pyridoxine as indicated.
First line IV glucose infusion with normal saline as needed for shock.
Second line Bicarbonate therapy for pH <7.0 and hemodialysis when indicated.< />pan>
Follow-up and disposition
Admission criteria All infants and children with suspected inborn errors of metabolism, significant ketosis, inability to tolerate oral intake, altered mental status, severe or persistent acidosis, refractory hypoglycemia, or hyperammonemia; ICU care for severe presentations. Transfer to a specialized pediatric center is often required.
Discharge criteria Normal mental status, adequate hydration, reassuring laboratory results, no significant intercurrent illness, and close follow-up arranged.
Follow-up recommendations Ongoing care with a metabolic disease specialist and primary care physician.
Pearls and pitfalls Always consider metabolic disease in unexplained pediatric decompensation. Do not delay glucose infusion. Avoid lactated Ringer solution. Use bicarbonate cautiously and only for severe acidosis. Early dialysis can be lifesaving in hyperammonemia.
Basics
Description Inborn errors of metabolism are inherited defects in metabolic pathways that lead to abnormal accumulation or deficiency of metabolites. More than 400 disorders are described, most presenting in infancy or childhood with variable and often nonspecific findings. Incidence ranges from 1:10,000 to 1:200,000 births. Major categories include amino acid disorders, urea cycle defects, organic acidemias, fatty acid oxidation defects, mitochondrial disorders, carbohydrate disorders, lysosomal storage diseases, peroxisomal disorders, and disorders of protein glycosylation. Pathophysiology relates directly to enzymatic or transport defects within metabolic pathways.
Etiology Genetic deficiency of enzymes or membrane transport systems involved in intermediary metabolism.
Diagnosis
Signs and symptoms Presentation may be acute and catastrophic or chronic and indolent. Neonates may initially be asymptomatic or present with hypothermia (mitochondrial defects), hypotonia or hypertonia (peroxisomal disorders), apnea (urea cycle defects, organic acidemias), seizures, vomiting, poor feeding, jaundice, hypoglycemia, dysmorphic features, or coma. Older untreated children may present with failure to thrive, dehydration, recurrent vomiting or diarrhea, food intolerance, lethargy, ataxia, seizures, or developmental delay.
History Review newborn screening results, dietary history, family history, consanguinity, and intercurrent illness.
Physical exam May reveal abnormal odor, altered mental status, tachypnea, dysmorphic facies, cataracts, cardiomyopathy, hepatosplenomegaly, dermatitis, or jaundice.
Essential workup Consider in any child with unexplained neurologic deterioration, persistent vomiting, metabolic acidosis, hypoglycemia, shock unresponsive to resuscitation, or failure to thrive.
Diagnosis tests and interpretation
Lab Bedside glucose, electrolytes, BUN/creatinine, CBC, calcium, liver function tests, bilirubin, coagulation studies, blood gas, lactate, pyruvate, uric acid, urinalysis, ammonia, serum amino acids, urine organic acids, and cultures as indicated.
Imaging CT head for altered mental status; chest radiograph as indicated.
Procedures Lumbar puncture when infection is a concern.
Differential diagnosis Sepsis, meningitis, encephalitis, dehydration, toxic ingestion, nonaccidental trauma, Reye syndrome, hepatic encephalopathy, endocrine disorders, renal failure, renal tubular acidosis, CNS mass lesions.
Treatment
Prehospital Prioritize ABCs and bedside glucose. Start IV glucose immediately; do not delay for fluids unless in shock. Avoid lactated Ringer solution. Keep patient NPO.
Initial stabilization Administer glucose (after Accu-Chek and thiamine when appropriate) and naloxone for altered mental status.
Emergency department management Secure airway, breathing, and circulation. Use normal saline for fluid boluses and avoid hypotonic fluids and lactated Ringer. Initiate IV glucose at 8–10 mg/kg/min (D10 at 1.5× maintenance) to prevent catabolism. Treat severe hypoglycemia with D25 bolus. Correct dehydration and acid–base abnormalities; consider bicarbonate only if pH <7.0. initiate dialysis for refractory severe acidosis or hyperammonemia. stop all oral intake initially. treat hyperammonemia urgently with ammonia-scavenging agents (arginine, sodium benzoate, phenylacetate, phenylbutyrate) in consultation a metabolic specialist. identify and precipitating infections.< />pan>
Medication D25 2–4 mL/kg IV for hypoglycemia, sodium bicarbonate 1–2 mEq/kg IV for severe acidosis, disease-specific therapies such as levocarnitine or pyridoxine as indicated.
First line IV glucose infusion with normal saline as needed for shock.
Second line Bicarbonate therapy for pH <7.0 and hemodialysis when indicated.< />pan>
Follow-up and disposition
Admission criteria All infants and children with suspected inborn errors of metabolism, significant ketosis, inability to tolerate oral intake, altered mental status, severe or persistent acidosis, refractory hypoglycemia, or hyperammonemia; ICU care for severe presentations. Transfer to a specialized pediatric center is often required.
Discharge criteria Normal mental status, adequate hydration, reassuring laboratory results, no significant intercurrent illness, and close follow-up arranged.
Follow-up recommendations Ongoing care with a metabolic disease specialist and primary care physician.
Pearls and pitfalls Always consider metabolic disease in unexplained pediatric decompensation. Do not delay glucose infusion. Avoid lactated Ringer solution. Use bicarbonate cautiously and only for severe acidosis. Early dialysis can be lifesaving in hyperammonemia.

- Published on
Emergency And Acute Medicine – Impetigo
Basics
Description Impetigo is a common superficial bacterial skin infection. It may be a primary infection of minor skin breaks or a secondary infection of preexisting dermatoses (impetiginization). It is most prevalent in children aged 2–5 years and occurs more often in warm, humid climates and summer months. Predisposing factors include minor trauma (especially around the nose), insect bites, burns, diabetes mellitus, HIV infection, varicella, and chronic skin disease. Complications include acute poststreptococcal glomerulonephritis (1–5% of nonbullous cases), cellulitis, sepsis, endocarditis, toxic shock syndrome, and staphylococcal scalded skin syndrome.
Etiology
Classic (nonbullous) impetigo results from bacterial entry through disrupted skin and is caused by Staphylococcus aureus, group A β-hemolytic streptococci, or both. Bullous impetigo is caused exclusively by S. aureus producing exfoliative toxins (A, B, D) that cleave desmoglein 1, leading to intraepidermal blistering.
Diagnosis
Signs and symptoms Nonbullous impetigo begins as erythematous macules or papules that evolve into vesicles or pustules, which rupture and form characteristic honey-colored crusts. Lesions are highly contagious and often pruritic. Regional lymphadenopathy may occur; systemic symptoms are uncommon. Bullous impetigo presents with flaccid bullae containing clear or yellow fluid that rupture easily, leaving erythematous bases; Nikolsky sign is absent. Fever and systemic symptoms are rare.
Physical exam Lesions most commonly involve the face, extremities, and scalp. Diagnosis is clinical based on appearance and distribution.
Essential workup
Routine laboratory testing is not required. Consider bacterial culture of bullae or pustules for refractory disease, outbreak investigation, or suspected methicillin-resistant S. aureus.
Diagnosis tests and interpretation
Lab Anti–DNase B titers may be elevated in poststreptococcal glomerulonephritis. Urinalysis is indicated if hematuria or edema suggests nephritis.
Imaging and procedures Not routinely indicated; biopsy is rarely necessary.
Differential diagnosis
Herpes simplex, varicella zoster, atopic or contact dermatitis, dermatophytosis, candidiasis, scabies, folliculitis, erysipelas, bullous pemphigoid, pemphigus vulgaris, staphylococcal scalded skin syndrome, Stevens–Johnson syndrome, thermal burns, toxic epidermal necrolysis, and cutaneous anthrax.
Treatment
Prehospital Cover lesions and use universal precautions to prevent transmission.
Initial stabilization Impetigo is rarely life-threatening in otherwise healthy patients.
Emergency department management Local care includes gentle cleansing, removal of crusts, and wet dressings. Small, localized nonbullous lesions may be treated with topical antibiotics alone. Extensive disease, bullous impetigo, lymphadenopathy, systemic symptoms, or outbreak settings require systemic antibiotics. Empiric therapy should cover S. aureus and streptococci; adjust for MRSA based on local resistance patterns.
Medication
Topical (nonbullous only): mupirocin 2% TID for 10 days or retapamulin 1% BID for 5 days.
Oral options (10 days unless noted): amoxicillin–clavulanate, cephalexin, dicloxacillin, clindamycin, azithromycin (5-day course), or trimethoprim–sulfamethoxazole when MRSA suspected. Doxycycline may be used for MRSA in patients ≥8 years.
Follow-up and disposition
Admission criteria Rare; consider for widespread disease, extensive bullae or denuded skin, dehydration, treatment failure, toxic appearance, immunocompromise, or neonates.
Discharge criteria Nontoxic patients with reliable caregivers and ability to comply with therapy.
Follow-up recommendations Primary care follow-up to ensure resolution. Return for lack of improvement or signs of nephritis (hematuria, periorbital or leg edema).
Pearls and pitfalls
Use systemic antibiotics for bullous disease or lymphadenopathy. Rising antibiotic resistance limits older regimens; suspect mupirocin resistance with treatment failure and switch agents. Treat affected household contacts simultaneously to prevent reinfection.
Basics
Description Impetigo is a common superficial bacterial skin infection. It may be a primary infection of minor skin breaks or a secondary infection of preexisting dermatoses (impetiginization). It is most prevalent in children aged 2–5 years and occurs more often in warm, humid climates and summer months. Predisposing factors include minor trauma (especially around the nose), insect bites, burns, diabetes mellitus, HIV infection, varicella, and chronic skin disease. Complications include acute poststreptococcal glomerulonephritis (1–5% of nonbullous cases), cellulitis, sepsis, endocarditis, toxic shock syndrome, and staphylococcal scalded skin syndrome.
Etiology
Classic (nonbullous) impetigo results from bacterial entry through disrupted skin and is caused by Staphylococcus aureus, group A β-hemolytic streptococci, or both. Bullous impetigo is caused exclusively by S. aureus producing exfoliative toxins (A, B, D) that cleave desmoglein 1, leading to intraepidermal blistering.
Diagnosis
Signs and symptoms Nonbullous impetigo begins as erythematous macules or papules that evolve into vesicles or pustules, which rupture and form characteristic honey-colored crusts. Lesions are highly contagious and often pruritic. Regional lymphadenopathy may occur; systemic symptoms are uncommon. Bullous impetigo presents with flaccid bullae containing clear or yellow fluid that rupture easily, leaving erythematous bases; Nikolsky sign is absent. Fever and systemic symptoms are rare.
Physical exam Lesions most commonly involve the face, extremities, and scalp. Diagnosis is clinical based on appearance and distribution.
Essential workup
Routine laboratory testing is not required. Consider bacterial culture of bullae or pustules for refractory disease, outbreak investigation, or suspected methicillin-resistant S. aureus.
Diagnosis tests and interpretation
Lab Anti–DNase B titers may be elevated in poststreptococcal glomerulonephritis. Urinalysis is indicated if hematuria or edema suggests nephritis.
Imaging and procedures Not routinely indicated; biopsy is rarely necessary.
Differential diagnosis
Herpes simplex, varicella zoster, atopic or contact dermatitis, dermatophytosis, candidiasis, scabies, folliculitis, erysipelas, bullous pemphigoid, pemphigus vulgaris, staphylococcal scalded skin syndrome, Stevens–Johnson syndrome, thermal burns, toxic epidermal necrolysis, and cutaneous anthrax.
Treatment
Prehospital Cover lesions and use universal precautions to prevent transmission.
Initial stabilization Impetigo is rarely life-threatening in otherwise healthy patients.
Emergency department management Local care includes gentle cleansing, removal of crusts, and wet dressings. Small, localized nonbullous lesions may be treated with topical antibiotics alone. Extensive disease, bullous impetigo, lymphadenopathy, systemic symptoms, or outbreak settings require systemic antibiotics. Empiric therapy should cover S. aureus and streptococci; adjust for MRSA based on local resistance patterns.
Medication
Topical (nonbullous only): mupirocin 2% TID for 10 days or retapamulin 1% BID for 5 days.
Oral options (10 days unless noted): amoxicillin–clavulanate, cephalexin, dicloxacillin, clindamycin, azithromycin (5-day course), or trimethoprim–sulfamethoxazole when MRSA suspected. Doxycycline may be used for MRSA in patients ≥8 years.
Follow-up and disposition
Admission criteria Rare; consider for widespread disease, extensive bullae or denuded skin, dehydration, treatment failure, toxic appearance, immunocompromise, or neonates.
Discharge criteria Nontoxic patients with reliable caregivers and ability to comply with therapy.
Follow-up recommendations Primary care follow-up to ensure resolution. Return for lack of improvement or signs of nephritis (hematuria, periorbital or leg edema).
Pearls and pitfalls
Use systemic antibiotics for bullous disease or lymphadenopathy. Rising antibiotic resistance limits older regimens; suspect mupirocin resistance with treatment failure and switch agents. Treat affected household contacts simultaneously to prevent reinfection.

- Published on
Emergency and Acute Medicine – Isopropanol poisoning
Isopropanol is a central nervous system depressant that is two to three times more potent than ethanol. It is rapidly absorbed after ingestion and is metabolized by alcohol dehydrogenase to acetone, which is also a CNS depressant. Unlike other toxic alcohols, isopropanol is ketogenic but does not cause a significant metabolic acidosis. Acetone is eliminated by the lungs and kidneys. The half-life of isopropanol ranges from 3 to 16 hours, while acetone has a longer half-life of 7.5 to 26 hours. Concomitant ethanol ingestion prolongs the half-life of isopropanol but not acetone.
Isopropanol is a clear, colorless, volatile liquid with a faint acetone odor and bitter taste, commonly found as 70% rubbing alcohol. It is present in many household and industrial products including disinfectants, hand sanitizers, solvents, window cleaners, paint removers, detergents, antifreeze, and toiletries. The typical adult patient is a chronic alcoholic who ingests isopropanol after depleting ethanol supplies. Dermal, inhalational, and rectal exposures can also cause systemic toxicity. Accidental ingestions are common in young children.
Symptoms usually develop within 30 to 60 minutes of exposure. Neurologic manifestations include lethargy, inebriation, vertigo, ataxia, headache, weakness, respiratory depression, coma, and apnea, without the initial excitatory phase seen with ethanol. Gastrointestinal findings include nausea, vomiting, abdominal pain, gastritis, and hematemesis. Cardiovascular effects include hypotension, tachycardia, myocardial depression, and peripheral vasodilation. Pulmonary findings include respiratory depression and hemorrhagic tracheobronchitis. Skin and eye exposure may cause irritation or chemical burns.
Diagnosis is based on history of exposure and the characteristic odor of isopropanol or acetone on the breath. Laboratory evaluation typically shows ketosis without significant acidosis unless end-organ hypoperfusion is present. Hypoglycemia may occur, particularly in children. Acetone can falsely elevate serum creatinine, which normalizes as acetone is metabolized. Urinalysis shows ketones, and an elevated osmolar gap is common. Coma is associated with serum isopropanol levels greater than 150 mg/dL. Imaging is guided by clinical findings, with chest radiography used to assess for aspiration and head CT considered for possible trauma.
Management is primarily supportive, with attention to airway protection, breathing, and circulation. Hypotension is treated with intravenous fluids and vasopressors if needed. There is no specific antidote, and treatment with ethanol or fomepizole is contraindicated. Activated charcoal may be considered if coingestants are suspected. Skin and eye exposures require copious irrigation. Hemodialysis effectively removes isopropanol and acetone and is reserved for patients with refractory hypotension, severe toxicity, or serum levels greater than 400 mg/dL.
Patients with moderate to severe toxicity, including altered mental status or hypotension, require hospital admission. Asymptomatic or mildly intoxicated patients may be observed for 2 to 4 hours and discharged if symptoms resolve. Referral for alcohol detoxification or psychiatric evaluation is recommended for intentional ingestions.
Supportive care remains the cornerstone of treatment, and a key pitfall is inappropriate use of ethanol or fomepizole, which should be avoided in isopropanol poisoning.
Isopropanol is a central nervous system depressant that is two to three times more potent than ethanol. It is rapidly absorbed after ingestion and is metabolized by alcohol dehydrogenase to acetone, which is also a CNS depressant. Unlike other toxic alcohols, isopropanol is ketogenic but does not cause a significant metabolic acidosis. Acetone is eliminated by the lungs and kidneys. The half-life of isopropanol ranges from 3 to 16 hours, while acetone has a longer half-life of 7.5 to 26 hours. Concomitant ethanol ingestion prolongs the half-life of isopropanol but not acetone.
Isopropanol is a clear, colorless, volatile liquid with a faint acetone odor and bitter taste, commonly found as 70% rubbing alcohol. It is present in many household and industrial products including disinfectants, hand sanitizers, solvents, window cleaners, paint removers, detergents, antifreeze, and toiletries. The typical adult patient is a chronic alcoholic who ingests isopropanol after depleting ethanol supplies. Dermal, inhalational, and rectal exposures can also cause systemic toxicity. Accidental ingestions are common in young children.
Symptoms usually develop within 30 to 60 minutes of exposure. Neurologic manifestations include lethargy, inebriation, vertigo, ataxia, headache, weakness, respiratory depression, coma, and apnea, without the initial excitatory phase seen with ethanol. Gastrointestinal findings include nausea, vomiting, abdominal pain, gastritis, and hematemesis. Cardiovascular effects include hypotension, tachycardia, myocardial depression, and peripheral vasodilation. Pulmonary findings include respiratory depression and hemorrhagic tracheobronchitis. Skin and eye exposure may cause irritation or chemical burns.
Diagnosis is based on history of exposure and the characteristic odor of isopropanol or acetone on the breath. Laboratory evaluation typically shows ketosis without significant acidosis unless end-organ hypoperfusion is present. Hypoglycemia may occur, particularly in children. Acetone can falsely elevate serum creatinine, which normalizes as acetone is metabolized. Urinalysis shows ketones, and an elevated osmolar gap is common. Coma is associated with serum isopropanol levels greater than 150 mg/dL. Imaging is guided by clinical findings, with chest radiography used to assess for aspiration and head CT considered for possible trauma.
Management is primarily supportive, with attention to airway protection, breathing, and circulation. Hypotension is treated with intravenous fluids and vasopressors if needed. There is no specific antidote, and treatment with ethanol or fomepizole is contraindicated. Activated charcoal may be considered if coingestants are suspected. Skin and eye exposures require copious irrigation. Hemodialysis effectively removes isopropanol and acetone and is reserved for patients with refractory hypotension, severe toxicity, or serum levels greater than 400 mg/dL.
Patients with moderate to severe toxicity, including altered mental status or hypotension, require hospital admission. Asymptomatic or mildly intoxicated patients may be observed for 2 to 4 hours and discharged if symptoms resolve. Referral for alcohol detoxification or psychiatric evaluation is recommended for intentional ingestions.
Supportive care remains the cornerstone of treatment, and a key pitfall is inappropriate use of ethanol or fomepizole, which should be avoided in isopropanol poisoning.
- Published on
Infectious Diseases and Microbiology: Ear Pain
Basics
Description
Otalgia denotes pain originating in or perceived in the ear. Evaluation should consider patient age and associated symptoms such as sore throat, fever, headache, visual changes, and symptom duration. A systematic ear examination is essential, beginning with the auricle and external auditory meatus, followed by inspection of the auditory canal and tympanic membrane to guide the differential diagnosis. A complete head and neck examination may uncover lymphadenopathy, pharyngeal or nasal inflammation, thyroid disease, or dental and oral pathology. Cerumen can obstruct visualization of the tympanic membrane and should be gently removed when necessary. Any exudate within the auditory canal should be cultured. Pain elicited by movement of the pinna suggests otitis externa, foreign body, or impacted cerumen. Perichondritis, an infectious process of the outer ear cartilage, must be differentiated from relapsing polychondritis, a noninfectious rheumatologic condition.
Epidemiology
Approximately half of ear pain cases are referred from non-otologic sources. Acute otitis externa, commonly termed swimmer’s ear, occurs more frequently during summer months. Recurrent acute otitis media affects about one-fifth of children. Head and neck malignancies typically occur after age 50 but may present earlier, even without classic risk factors; several forms of nasopharyngeal carcinoma are associated with Epstein–Barr virus infection. Auricular cellulitis often follows minor trauma. Perichondritis usually develops after burns, trauma, or upper-ear piercings and is most commonly caused by Pseudomonas aeruginosa or Staphylococcus aureus. Chronic otitis externa often results from repeated minor trauma such as scratching or cotton swab use, and chronic middle-ear drainage may be mistaken for this condition. Malignant otitis externa is a destructive infection of the external canal and skull base.
Etiology
Ear pain is frequently referred because the ear receives sensory innervation from cranial nerves V, VII, VIII, IX, and X. Referred otalgia may arise from dental disease, gingival abscesses, nasopharyngeal or laryngeal inflammation or tumors, sinusitis, temporomandibular joint disorders, tonsillitis, tongue lesions, cervical spine disease, neural irritation such as trigeminal neuralgia or acoustic neuroma, gastroesophageal reflux in infants, thyroiditis, lateral sinus thrombosis, posterior fossa inflammation, or medication effects. Primary ear pathology causing otalgia includes acute or chronic otitis media, tympanic membrane rupture, anterior canal wall fracture, mastoiditis, Ménière disease, and eustachian tube dysfunction. Malignant otitis externa is almost always due to Pseudomonas aeruginosa. Otalgia may also accompany migraine, atypical facial pain, and herpes simplex infection of cranial nerves V, VII, or IX. Herpes zoster affecting the external auditory canal may produce ipsilateral facial paralysis, known as Ramsay Hunt syndrome, from involvement of the geniculate ganglion. Facial nerve palsy may also occur with Lyme disease. Neoplasms of the infratemporal fossa may present solely with ear pain. Acute otitis externa is most often caused by Pseudomonas aeruginosa, Staphylococcus aureus, or streptococcal species, with swimming and canal trauma as major risk factors.
Diagnosis
Clinical Manifestations
Auricular cellulitis presents with a swollen, erythematous, warm, mildly tender ear. Perichondritis causes marked swelling, redness, heat, and severe tenderness of the pinna with relative sparing of the lobule. Chronic otitis externa more commonly causes itching than pain. Nasopharyngeal carcinoma may be asymptomatic early but often produces unilateral serous otitis media from eustachian tube obstruction, nasal blockage, or epistaxis, and advanced disease may cause cranial nerve palsies, especially involving nerves III, IV, VI, and VII. Malignant otitis externa typically affects elderly patients with diabetes or individuals with HIV and presents with severe otalgia, otorrhea, possible hearing loss, tender pinna, trismus from temporomandibular involvement, and sometimes cranial nerve palsies, most often of nerve VII. Fever and weight loss are uncommon. Examination reveals canal edema, erythema, purulent discharge, debris, and granulation tissue. Vesicular lesions in the external canal suggest herpes zoster and warrant evaluation for facial nerve palsy. Hearing loss with abnormal tympanic membrane findings indicates serous or bacterial otitis media or cholesteatoma. Nasal polyps, marked septal deviation, or nasopharyngeal tumors may be associated with otitis media.
Physical Examination
Findings such as an erythematous canal with discharge, preauricular lymphadenopathy, and pain on tragal or pinna manipulation suggest otitis externa. Fever, irritability in children, and a bulging or erythematous tympanic membrane with loss of the cone of light indicate otitis media, sometimes with canal pus if perforation is present. Altered mental status or meningeal signs such as headache and neck stiffness indicate possible central nervous system involvement and require urgent management.
Diagnostic Tests and Interpretation
Laboratory Studies
Peripheral leukocytosis is uncommon in malignant otitis externa, whereas erythrocyte sedimentation rate is typically elevated. Cerebrospinal fluid analysis may occasionally show pleocytosis and increased protein.
Imaging
Dental pathology can be assessed with panoramic radiography. In malignant otitis externa, CT of the temporal bone or mastoid often demonstrates bony erosion and new bone formation, while MRI more accurately defines soft-tissue extension and skull-base involvement.
Treatment
Medications
Acute otitis externa is managed with careful canal cleaning and topical therapy using antiseptics or antibiotic drops such as polymyxin–neomycin, along with counseling on ear hygiene and water avoidance. Malignant otitis externa requires urgent otolaryngology consultation and prolonged antipseudomonal therapy with agents such as cefepime, ceftazidime, carbapenems, or fluoroquinolones, typically for at least three to four weeks and longer if bone involvement is present. Auricular cellulitis is treated with warm compresses and intravenous antibiotics targeting staphylococci and streptococci. Severe perichondritis requires extended antibiotic therapy, often with agents such as piperacillin–tazobactam or nafcillin combined with ciprofloxacin, and may benefit from incision and drainage. Ramsay Hunt syndrome is treated with acyclovir and corticosteroids, and early therapy improves facial nerve outcomes; ophthalmologic evaluation is recommended to assess ocular involvement.
Ongoing Care and Follow-Up
Referral to an otolaryngologist is advised when ear pain persists despite appropriate initial evaluation and management.
Complications
Otitis media can lead to mastoiditis, epidural abscess, dural venous sinus thrombosis, meningitis, or brain abscess. Malignant otitis externa may extend to the cavernous sinus or contralateral petrous apex, and although meningitis and brain abscess are uncommon, they are serious potential sequelae.
Basics
Description
Otalgia denotes pain originating in or perceived in the ear. Evaluation should consider patient age and associated symptoms such as sore throat, fever, headache, visual changes, and symptom duration. A systematic ear examination is essential, beginning with the auricle and external auditory meatus, followed by inspection of the auditory canal and tympanic membrane to guide the differential diagnosis. A complete head and neck examination may uncover lymphadenopathy, pharyngeal or nasal inflammation, thyroid disease, or dental and oral pathology. Cerumen can obstruct visualization of the tympanic membrane and should be gently removed when necessary. Any exudate within the auditory canal should be cultured. Pain elicited by movement of the pinna suggests otitis externa, foreign body, or impacted cerumen. Perichondritis, an infectious process of the outer ear cartilage, must be differentiated from relapsing polychondritis, a noninfectious rheumatologic condition.
Epidemiology
Approximately half of ear pain cases are referred from non-otologic sources. Acute otitis externa, commonly termed swimmer’s ear, occurs more frequently during summer months. Recurrent acute otitis media affects about one-fifth of children. Head and neck malignancies typically occur after age 50 but may present earlier, even without classic risk factors; several forms of nasopharyngeal carcinoma are associated with Epstein–Barr virus infection. Auricular cellulitis often follows minor trauma. Perichondritis usually develops after burns, trauma, or upper-ear piercings and is most commonly caused by Pseudomonas aeruginosa or Staphylococcus aureus. Chronic otitis externa often results from repeated minor trauma such as scratching or cotton swab use, and chronic middle-ear drainage may be mistaken for this condition. Malignant otitis externa is a destructive infection of the external canal and skull base.
Etiology
Ear pain is frequently referred because the ear receives sensory innervation from cranial nerves V, VII, VIII, IX, and X. Referred otalgia may arise from dental disease, gingival abscesses, nasopharyngeal or laryngeal inflammation or tumors, sinusitis, temporomandibular joint disorders, tonsillitis, tongue lesions, cervical spine disease, neural irritation such as trigeminal neuralgia or acoustic neuroma, gastroesophageal reflux in infants, thyroiditis, lateral sinus thrombosis, posterior fossa inflammation, or medication effects. Primary ear pathology causing otalgia includes acute or chronic otitis media, tympanic membrane rupture, anterior canal wall fracture, mastoiditis, Ménière disease, and eustachian tube dysfunction. Malignant otitis externa is almost always due to Pseudomonas aeruginosa. Otalgia may also accompany migraine, atypical facial pain, and herpes simplex infection of cranial nerves V, VII, or IX. Herpes zoster affecting the external auditory canal may produce ipsilateral facial paralysis, known as Ramsay Hunt syndrome, from involvement of the geniculate ganglion. Facial nerve palsy may also occur with Lyme disease. Neoplasms of the infratemporal fossa may present solely with ear pain. Acute otitis externa is most often caused by Pseudomonas aeruginosa, Staphylococcus aureus, or streptococcal species, with swimming and canal trauma as major risk factors.
Diagnosis
Clinical Manifestations
Auricular cellulitis presents with a swollen, erythematous, warm, mildly tender ear. Perichondritis causes marked swelling, redness, heat, and severe tenderness of the pinna with relative sparing of the lobule. Chronic otitis externa more commonly causes itching than pain. Nasopharyngeal carcinoma may be asymptomatic early but often produces unilateral serous otitis media from eustachian tube obstruction, nasal blockage, or epistaxis, and advanced disease may cause cranial nerve palsies, especially involving nerves III, IV, VI, and VII. Malignant otitis externa typically affects elderly patients with diabetes or individuals with HIV and presents with severe otalgia, otorrhea, possible hearing loss, tender pinna, trismus from temporomandibular involvement, and sometimes cranial nerve palsies, most often of nerve VII. Fever and weight loss are uncommon. Examination reveals canal edema, erythema, purulent discharge, debris, and granulation tissue. Vesicular lesions in the external canal suggest herpes zoster and warrant evaluation for facial nerve palsy. Hearing loss with abnormal tympanic membrane findings indicates serous or bacterial otitis media or cholesteatoma. Nasal polyps, marked septal deviation, or nasopharyngeal tumors may be associated with otitis media.
Physical Examination
Findings such as an erythematous canal with discharge, preauricular lymphadenopathy, and pain on tragal or pinna manipulation suggest otitis externa. Fever, irritability in children, and a bulging or erythematous tympanic membrane with loss of the cone of light indicate otitis media, sometimes with canal pus if perforation is present. Altered mental status or meningeal signs such as headache and neck stiffness indicate possible central nervous system involvement and require urgent management.
Diagnostic Tests and Interpretation
Laboratory Studies
Peripheral leukocytosis is uncommon in malignant otitis externa, whereas erythrocyte sedimentation rate is typically elevated. Cerebrospinal fluid analysis may occasionally show pleocytosis and increased protein.
Imaging
Dental pathology can be assessed with panoramic radiography. In malignant otitis externa, CT of the temporal bone or mastoid often demonstrates bony erosion and new bone formation, while MRI more accurately defines soft-tissue extension and skull-base involvement.
Treatment
Medications
Acute otitis externa is managed with careful canal cleaning and topical therapy using antiseptics or antibiotic drops such as polymyxin–neomycin, along with counseling on ear hygiene and water avoidance. Malignant otitis externa requires urgent otolaryngology consultation and prolonged antipseudomonal therapy with agents such as cefepime, ceftazidime, carbapenems, or fluoroquinolones, typically for at least three to four weeks and longer if bone involvement is present. Auricular cellulitis is treated with warm compresses and intravenous antibiotics targeting staphylococci and streptococci. Severe perichondritis requires extended antibiotic therapy, often with agents such as piperacillin–tazobactam or nafcillin combined with ciprofloxacin, and may benefit from incision and drainage. Ramsay Hunt syndrome is treated with acyclovir and corticosteroids, and early therapy improves facial nerve outcomes; ophthalmologic evaluation is recommended to assess ocular involvement.
Ongoing Care and Follow-Up
Referral to an otolaryngologist is advised when ear pain persists despite appropriate initial evaluation and management.
Complications
Otitis media can lead to mastoiditis, epidural abscess, dural venous sinus thrombosis, meningitis, or brain abscess. Malignant otitis externa may extend to the cavernous sinus or contralateral petrous apex, and although meningitis and brain abscess are uncommon, they are serious potential sequelae.
- Published on
Emergency And Acute Medicine – Knee Dislocation
Basics Description
Knee dislocation is defined by displacement of the tibia relative to the distal femur and represents a true orthopedic emergency because of the high risk of vascular and neurologic injury. Anterior dislocation is the most common type, accounting for approximately 60% of cases, and typically results from hyperextension. Rupture of the posterior capsule may occur at 30° of hyperextension, with disruption of the posterior cruciate ligament and potential popliteal artery injury at greater degrees. Posterior dislocation usually results from a direct blow to the anterior tibia with the knee flexed, such as a dashboard injury. Medial and lateral dislocations occur from varus or valgus stress, respectively, and are associated with multiligamentous injuries.
Popliteal artery injury occurs in up to 35% of knee dislocations and is the most limb-threatening complication. Anterior dislocations commonly cause traction-related intimal injury with delayed thrombosis, whereas posterior dislocations are more likely to cause direct arterial transection and immediate thrombosis. Peroneal nerve injury is less common but clinically significant and often accompanies arterial injury, particularly in medial or rotational dislocations.
Etiology
Knee dislocations typically result from high-energy mechanisms such as motor vehicle collisions, auto–pedestrian trauma, or athletic injuries, most commonly football.
Diagnosis Signs And Symptoms
Patients may present with a grossly deformed or unstable knee, though spontaneous reduction can mask deformity. Absence of distal pulses or signs of ischemia such as pallor, paresthesia, pain, paralysis, or poikilothermia raise immediate concern for popliteal artery injury. Neurologic deficits may include decreased sensation in the first web space or inability to dorsiflex the foot. A careful history of the mechanism of injury and repeated neurovascular examinations are essential.
Essential Workup
Evaluation requires a detailed history and thorough physical examination with emphasis on vascular and neurologic status. Distal pulses should be assessed by palpation and Doppler, capillary refill documented, and ankle–brachial index calculated. Neurologic examination should assess peroneal nerve function. Repeat examinations are mandatory, especially after reduction.
Diagnosis Tests And Interpretation
AP and lateral knee radiographs are required to confirm alignment and exclude associated fractures. MRI within one week is useful for defining ligamentous injury. An ABI ≥0.9 suggests low likelihood of major arterial injury, but abnormal findings warrant further evaluation. Vascular ultrasound or arteriography should be considered when pulses are diminished, ischemic symptoms persist, or peroneal nerve injury is present.
Differential Diagnosis
Conditions to consider include tibial plateau fracture, supracondylar femoral fracture, and ligamentous or tendinous avulsion injuries.
Treatment Pre Hospital
Initial management focuses on airway, breathing, and circulation. Distal pulses and motor function must be documented. The knee should be splinted in slight flexion to minimize popliteal artery tension.
Initial Stabilization Therapy
Address ABCs, particularly in polytrauma patients. Hypotension should be corrected, as it may obscure vascular compromise. Immediate closed reduction is indicated for any evidence of limb ischemia, with early orthopedic and vascular consultation.
Ed Treatment Procedures
Closed reduction is performed using longitudinal traction with gentle realignment, avoiding pressure in the popliteal fossa. The knee is immobilized in 15–20° of flexion. Neurovascular status must be reassessed immediately after reduction and monitored frequently. IV analgesia should be provided, and surgical consultation obtained for open injuries, vascular compromise, or irreducible dislocations.
Medication
IV narcotic analgesia is first-line. Oral medications should be avoided due to the high likelihood of operative intervention.
Follow-Up Disposition
All patients with knee dislocation require admission for observation and management of potential vascular injury. Discharge from the ED is not appropriate.
Follow-Up Recommendations
Orthopedic follow-up is required for staged ligamentous repair, typically delayed until swelling subsides. Vascular surgery follow-up is necessary if popliteal artery injury is identified.
Pearls And Pitfalls
Failure to restore popliteal artery flow within 6–8 hours carries an amputation risk approaching 90%. Peroneal nerve injury has a poor prognosis for recovery. Delayed compartment syndrome may occur and requires vigilant monitoring.
Basics Description
Knee dislocation is defined by displacement of the tibia relative to the distal femur and represents a true orthopedic emergency because of the high risk of vascular and neurologic injury. Anterior dislocation is the most common type, accounting for approximately 60% of cases, and typically results from hyperextension. Rupture of the posterior capsule may occur at 30° of hyperextension, with disruption of the posterior cruciate ligament and potential popliteal artery injury at greater degrees. Posterior dislocation usually results from a direct blow to the anterior tibia with the knee flexed, such as a dashboard injury. Medial and lateral dislocations occur from varus or valgus stress, respectively, and are associated with multiligamentous injuries.
Popliteal artery injury occurs in up to 35% of knee dislocations and is the most limb-threatening complication. Anterior dislocations commonly cause traction-related intimal injury with delayed thrombosis, whereas posterior dislocations are more likely to cause direct arterial transection and immediate thrombosis. Peroneal nerve injury is less common but clinically significant and often accompanies arterial injury, particularly in medial or rotational dislocations.
Etiology
Knee dislocations typically result from high-energy mechanisms such as motor vehicle collisions, auto–pedestrian trauma, or athletic injuries, most commonly football.
Diagnosis Signs And Symptoms
Patients may present with a grossly deformed or unstable knee, though spontaneous reduction can mask deformity. Absence of distal pulses or signs of ischemia such as pallor, paresthesia, pain, paralysis, or poikilothermia raise immediate concern for popliteal artery injury. Neurologic deficits may include decreased sensation in the first web space or inability to dorsiflex the foot. A careful history of the mechanism of injury and repeated neurovascular examinations are essential.
Essential Workup
Evaluation requires a detailed history and thorough physical examination with emphasis on vascular and neurologic status. Distal pulses should be assessed by palpation and Doppler, capillary refill documented, and ankle–brachial index calculated. Neurologic examination should assess peroneal nerve function. Repeat examinations are mandatory, especially after reduction.
Diagnosis Tests And Interpretation
AP and lateral knee radiographs are required to confirm alignment and exclude associated fractures. MRI within one week is useful for defining ligamentous injury. An ABI ≥0.9 suggests low likelihood of major arterial injury, but abnormal findings warrant further evaluation. Vascular ultrasound or arteriography should be considered when pulses are diminished, ischemic symptoms persist, or peroneal nerve injury is present.
Differential Diagnosis
Conditions to consider include tibial plateau fracture, supracondylar femoral fracture, and ligamentous or tendinous avulsion injuries.
Treatment Pre Hospital
Initial management focuses on airway, breathing, and circulation. Distal pulses and motor function must be documented. The knee should be splinted in slight flexion to minimize popliteal artery tension.
Initial Stabilization Therapy
Address ABCs, particularly in polytrauma patients. Hypotension should be corrected, as it may obscure vascular compromise. Immediate closed reduction is indicated for any evidence of limb ischemia, with early orthopedic and vascular consultation.
Ed Treatment Procedures
Closed reduction is performed using longitudinal traction with gentle realignment, avoiding pressure in the popliteal fossa. The knee is immobilized in 15–20° of flexion. Neurovascular status must be reassessed immediately after reduction and monitored frequently. IV analgesia should be provided, and surgical consultation obtained for open injuries, vascular compromise, or irreducible dislocations.
Medication
IV narcotic analgesia is first-line. Oral medications should be avoided due to the high likelihood of operative intervention.
Follow-Up Disposition
All patients with knee dislocation require admission for observation and management of potential vascular injury. Discharge from the ED is not appropriate.
Follow-Up Recommendations
Orthopedic follow-up is required for staged ligamentous repair, typically delayed until swelling subsides. Vascular surgery follow-up is necessary if popliteal artery injury is identified.
Pearls And Pitfalls
Failure to restore popliteal artery flow within 6–8 hours carries an amputation risk approaching 90%. Peroneal nerve injury has a poor prognosis for recovery. Delayed compartment syndrome may occur and requires vigilant monitoring.
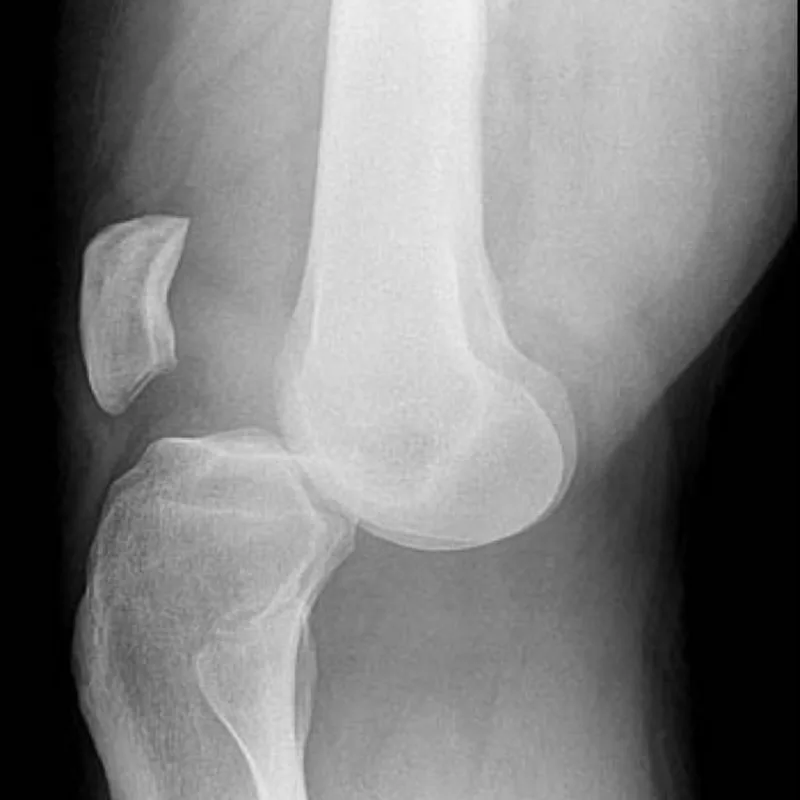
- Published on
Emergency and Acute Medicine – Kawasaki disease
Kawasaki disease is an acute, self-limited inflammatory illness involving multiple organ systems and is the leading cause of acquired heart disease in children in developed countries. It is a medium-vessel vasculitis with particular predilection for the coronary arteries. Acute cardiac complications include myocarditis, pericarditis, and coronary artery aneurysms, which may later progress to stenosis or thrombosis; giant aneurysms carry a risk of rupture. The disease progresses through three stages: an acute phase lasting 1–2 weeks characterized by fever and systemic inflammation; a subacute phase extending to approximately 4 weeks marked by desquamation, thrombocytosis, and the highest risk of sudden death from coronary involvement; and a convalescent phase lasting 6–8 weeks during which symptoms resolve and inflammatory markers normalize.
Epidemiologically, 80% of cases occur in children younger than 4 years, with a peak incidence between 1 and 2 years of age. The disease is rare in infants younger than 3 months, though adult cases have been reported. Males are affected more often than females, and children of Asian descent have the highest incidence, suggesting a genetic predisposition. Delayed diagnosis, prolonged fever, male sex, extremes of age, elevated inflammatory markers, anemia, hypoalbuminemia, and abnormal initial echocardiography increase the risk of coronary artery aneurysm formation. Approximately 10–15% of patients are resistant to standard therapy.
The etiology remains unknown, but the disease is thought to represent an abnormal immune response to an infectious trigger in genetically susceptible individuals. Seasonal clustering and epidemic patterns support this hypothesis, with increased incidence in winter and early spring.
Diagnosis is clinical. Classic Kawasaki disease is defined by fever lasting at least 5 days in combination with four of five features: bilateral nonexudative conjunctival injection, oral mucosal changes (such as strawberry tongue or cracked lips), polymorphous rash, extremity changes (erythema, edema, or desquamation), and cervical lymphadenopathy greater than 1.5 cm. Incomplete or atypical Kawasaki disease should be suspected in children with prolonged fever and fewer criteria accompanied by elevated ESR or CRP and supportive laboratory findings such as anemia, hypoalbuminemia, elevated ALT, leukocytosis, thrombocytosis after day 7, or sterile pyuria.
Clinical presentation typically includes abrupt onset of high, spiking fever persisting longer than 5 days. Children often appear irritable and ill. Ocular findings include conjunctivitis without exudate and photophobia. Oral findings include erythema, fissured lips, and strawberry tongue. Skin findings are variable and usually involve the trunk, while extremity changes include painful edema and erythema followed later by desquamation of the fingers and toes. Gastrointestinal symptoms such as vomiting, diarrhea, abdominal pain, and gallbladder hydrops may occur. Cardiac manifestations include myocarditis, pericarditis, heart failure, and, later, coronary artery aneurysms.
Evaluation requires a high index of suspicion in any febrile child with rash. Laboratory findings typically include elevated ESR and CRP, leukocytosis with left shift, normocytic anemia, thrombocytosis in the subacute phase, sterile pyuria, and mild transaminase elevation. Blood, urine, CSF, and throat cultures are negative. Echocardiography is essential to assess coronary artery involvement and should be performed at diagnosis, at 2–3 weeks, and again at 6–8 weeks. ECG may be indicated if myocardial involvement is suspected.
The differential diagnosis includes viral illnesses such as adenovirus, measles, Epstein–Barr virus, and influenza; bacterial infections such as scarlet fever, staphylococcal scalded skin syndrome, and rickettsial disease; immune-mediated conditions including Stevens–Johnson syndrome and serum sickness; and other vasculitides or connective tissue diseases.
Management focuses on early treatment to prevent cardiac complications. All patients meeting diagnostic criteria should be admitted. Initial therapy consists of intravenous immunoglobulin and high-dose aspirin, ideally within the first 10 days of illness, which reduces the incidence of coronary artery aneurysms from approximately 20–25% to 2–4%. Cardiology consultation is required, and myocardial infarction is treated similarly to adults if it occurs.
First-line treatment includes IVIG at 2 g/kg administered over 10–12 hours, with repeat dosing for persistent or recrudescent fever. High-dose aspirin is given during the acute phase for anti-inflammatory effect, followed by low-dose aspirin for antiplatelet therapy during the convalescent period. Patients who fail two doses of IVIG may require corticosteroids or other immunomodulatory therapies such as infliximab or cyclosporine.
All patients diagnosed with Kawasaki disease require hospitalization, while nontoxic children who do not meet diagnostic criteria may be discharged with close follow-up. Long-term cardiology follow-up is mandatory for monitoring coronary artery involvement.
Early recognition and treatment are critical, as prompt IVIG and aspirin therapy prevent coronary aneurysm formation in the vast majority of patients. Kawasaki disease should be strongly considered in children with persistent fever and repeated emergency visits, and incomplete presentations must not be overlooked.
Kawasaki disease is an acute, self-limited inflammatory illness involving multiple organ systems and is the leading cause of acquired heart disease in children in developed countries. It is a medium-vessel vasculitis with particular predilection for the coronary arteries. Acute cardiac complications include myocarditis, pericarditis, and coronary artery aneurysms, which may later progress to stenosis or thrombosis; giant aneurysms carry a risk of rupture. The disease progresses through three stages: an acute phase lasting 1–2 weeks characterized by fever and systemic inflammation; a subacute phase extending to approximately 4 weeks marked by desquamation, thrombocytosis, and the highest risk of sudden death from coronary involvement; and a convalescent phase lasting 6–8 weeks during which symptoms resolve and inflammatory markers normalize.
Epidemiologically, 80% of cases occur in children younger than 4 years, with a peak incidence between 1 and 2 years of age. The disease is rare in infants younger than 3 months, though adult cases have been reported. Males are affected more often than females, and children of Asian descent have the highest incidence, suggesting a genetic predisposition. Delayed diagnosis, prolonged fever, male sex, extremes of age, elevated inflammatory markers, anemia, hypoalbuminemia, and abnormal initial echocardiography increase the risk of coronary artery aneurysm formation. Approximately 10–15% of patients are resistant to standard therapy.
The etiology remains unknown, but the disease is thought to represent an abnormal immune response to an infectious trigger in genetically susceptible individuals. Seasonal clustering and epidemic patterns support this hypothesis, with increased incidence in winter and early spring.
Diagnosis is clinical. Classic Kawasaki disease is defined by fever lasting at least 5 days in combination with four of five features: bilateral nonexudative conjunctival injection, oral mucosal changes (such as strawberry tongue or cracked lips), polymorphous rash, extremity changes (erythema, edema, or desquamation), and cervical lymphadenopathy greater than 1.5 cm. Incomplete or atypical Kawasaki disease should be suspected in children with prolonged fever and fewer criteria accompanied by elevated ESR or CRP and supportive laboratory findings such as anemia, hypoalbuminemia, elevated ALT, leukocytosis, thrombocytosis after day 7, or sterile pyuria.
Clinical presentation typically includes abrupt onset of high, spiking fever persisting longer than 5 days. Children often appear irritable and ill. Ocular findings include conjunctivitis without exudate and photophobia. Oral findings include erythema, fissured lips, and strawberry tongue. Skin findings are variable and usually involve the trunk, while extremity changes include painful edema and erythema followed later by desquamation of the fingers and toes. Gastrointestinal symptoms such as vomiting, diarrhea, abdominal pain, and gallbladder hydrops may occur. Cardiac manifestations include myocarditis, pericarditis, heart failure, and, later, coronary artery aneurysms.
Evaluation requires a high index of suspicion in any febrile child with rash. Laboratory findings typically include elevated ESR and CRP, leukocytosis with left shift, normocytic anemia, thrombocytosis in the subacute phase, sterile pyuria, and mild transaminase elevation. Blood, urine, CSF, and throat cultures are negative. Echocardiography is essential to assess coronary artery involvement and should be performed at diagnosis, at 2–3 weeks, and again at 6–8 weeks. ECG may be indicated if myocardial involvement is suspected.
The differential diagnosis includes viral illnesses such as adenovirus, measles, Epstein–Barr virus, and influenza; bacterial infections such as scarlet fever, staphylococcal scalded skin syndrome, and rickettsial disease; immune-mediated conditions including Stevens–Johnson syndrome and serum sickness; and other vasculitides or connective tissue diseases.
Management focuses on early treatment to prevent cardiac complications. All patients meeting diagnostic criteria should be admitted. Initial therapy consists of intravenous immunoglobulin and high-dose aspirin, ideally within the first 10 days of illness, which reduces the incidence of coronary artery aneurysms from approximately 20–25% to 2–4%. Cardiology consultation is required, and myocardial infarction is treated similarly to adults if it occurs.
First-line treatment includes IVIG at 2 g/kg administered over 10–12 hours, with repeat dosing for persistent or recrudescent fever. High-dose aspirin is given during the acute phase for anti-inflammatory effect, followed by low-dose aspirin for antiplatelet therapy during the convalescent period. Patients who fail two doses of IVIG may require corticosteroids or other immunomodulatory therapies such as infliximab or cyclosporine.
All patients diagnosed with Kawasaki disease require hospitalization, while nontoxic children who do not meet diagnostic criteria may be discharged with close follow-up. Long-term cardiology follow-up is mandatory for monitoring coronary artery involvement.
Early recognition and treatment are critical, as prompt IVIG and aspirin therapy prevent coronary aneurysm formation in the vast majority of patients. Kawasaki disease should be strongly considered in children with persistent fever and repeated emergency visits, and incomplete presentations must not be overlooked.
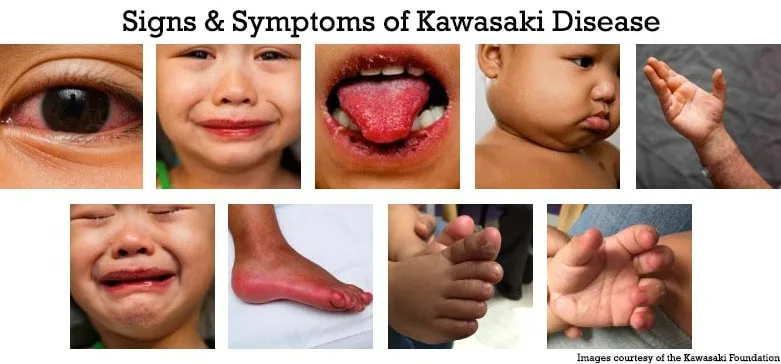
- Published on
Infectious Diseases and Microbiology: Genital Lesions and Ulcers
Basics
Description
Genital lesions involve the reproductive organs and can result from a range of infectious and noninfectious conditions.
Epidemiology
Genital ulcers are the most frequent sexually transmitted cause of genital lesions. In North America and Europe, the leading sexually transmitted causes of genital ulcer disease are herpes simplex and syphilis. In the United States, a substantial proportion of people aged 14–49 have HSV-2 infection, and HSV-1 accounts for a significant minority of genital herpes cases. Chancroid is more common in Africa, Asia, and Latin America but can occur in sporadic U.S. outbreaks. Lymphogranuloma venereum is uncommon in industrialized countries, though outbreaks have been reported among men who have sex with men.
Risk Factors
About half of exposed sexual partners may acquire genital warts (HPV) after contact with an infected partner. Sexual transmission of syphilis can be high, with rates reported up to roughly one-third per exposure.
General Prevention
Risk reduction includes abstinence, consistent condom use, limiting partners, safer-sex practices, and HPV vaccination. Two HPV vaccines are available for females aged 9–26 that protect against HPV 6 and 11 (responsible for most genital warts) and HPV 16 and 18 (responsible for a large proportion of cervical cancers): the quadrivalent vaccine (Gardasil) and the bivalent vaccine (Cervarix). Quadrivalent vaccination in males aged 9–26 can reduce genital warts.
Etiology
Infectious Ulcers
Common causes include genital herpes, syphilis due to Treponema pallidum, chancroid due to Haemophilus ducreyi, lymphogranuloma venereum from Chlamydia trachomatis serovars L1–L3, donovanosis (granuloma inguinale) from Klebsiella granulomatis, and less commonly tuberculosis or tularemia. Additional reported etiologies include candidiasis, histoplasmosis, amebiasis, gonorrhea, and trichomoniasis. Acute or primary HIV infection can present with genital ulcers, and a notable minority of ulcer cases involve more than one pathogen.
Noninfectious Ulcers
Noninfectious causes include trauma, fixed drug eruption, malignancy, systemic lupus erythematosus, and Behçet disease.
Other Lesion Patterns
Papules may be due to candidiasis, molluscum contagiosum, scabies, syphilis, or condylomata acuminata from HPV, with types 6 and 11 most common and types 16, 18, 31, 33, and 35 linked to cervical dysplasia. Vesicles or bullae suggest herpes, impetigo, or scabies. Diffuse erythema may occur with candidiasis, erysipelas often after trauma or surgery, contact dermatitis, drug eruption, psoriasis, or trauma. Other patterns include benign cysts, nodules from hidradenitis or furunculosis, and crusted lesions from herpes or scabies
.
Commonly Associated Conditions
HIV and other sexually transmitted infections commonly coexist with genital lesions.
Diagnosis
History
Key points include time of onset, associated symptoms, immunodeficiency, prior sexually transmitted infections, prior similar lesions, and travel. Typical incubation periods vary by cause, including herpes within days but sometimes up to weeks, genital warts usually weeks to months, syphilis typically weeks but up to months, chancroid usually within a week but variable, donovanosis over weeks, molluscum potentially months, and ectoparasites such as pubic lice or scabies often after several weeks. Lesions appearing within hours of exposure suggest trauma, chemical irritation, or hypersensitivity. Itching is common early in herpes and with scabies or pubic lice, and mild pruritus can occur in secondary syphilis.
Physical Examination
Define lesion morphology and distribution, determine whether disease is localized or generalized, assess tenderness, lymphadenopathy, and any urethral or vaginal discharge, and examine buccal mucosa and perianal skin; pelvic examination or prostate assessment is often needed. Early herpes often begins as clustered vesicles on an erythematous base, may show umbilication, is typically very painful, and is often ulcerated by presentation. Pain is typical of herpes, chancroid, and tularemia, whereas donovanosis ulcers are usually painless. Ulcer base and borders can help narrow the differential: Behçet ulcers may be yellow and necrotic, chancroid bases are often necrotic, donovanosis can appear beefy red with hypertrophy, and syphilitic and herpetic ulcers may have cleaner bases; chancroid ulcers are classically nonindurated with irregular erythematous edges, syphilitic ulcers are indurated, and herpetic ulcers often have erythematous borders. Primary syphilis more often produces a solitary lesion, while chancroid often causes multiple ulcers of varying size. Behçet disease can cause recurrent, multiple genital ulcers involving scrotum or vulva and may be accompanied by recurrent oral ulcers and skin or eye disease, which may not occur simultaneously. Linear burrows suggest scabies, and reddish specks may reflect crab louse excreta. Urethral discharge suggests gonorrhea or reactive arthritis. In adults with genital lesions, inguinal lymphadenopathy supports consideration of chancroid, LGV, syphilis, herpes, lymphoma, or tuberculosis. LGV ulcers may be unnoticed and heal spontaneously, followed weeks later by painful inguinal lymphadenopathy and systemic symptoms, and untreated disease can progress to elephantiasis-like genital distortion. Lymphadenopathy tends to be unilateral in chancroid and LGV and more often bilateral in genital herpes. Painful perianal ulcers or mucosal ulcers seen on anoscopy should be treated presumptively for HSV and LGV. Molluscum contagiosum causes mildly contagious umbilicated papules up to about 1 cm, and advanced HIV may cause widespread or large lesions. Tuberculous genital lesions may appear as chronic minimally painful red, firm, nodular sores. Clinical appearance alone is often insufficient for diagnosis.
Diagnostic Tests and Interpretation
Laboratory Studies
All patients with genital, anal, or perianal ulcers should receive syphilis serology with dark-field examination when available, HSV testing by culture or PCR or type-specific serology, and HIV testing. If suspicion and culture capacity exist, obtain H. ducreyi culture for chancroid. Donovanosis can be supported by identifying intracellular Donovan bodies on Giemsa or Wright stain from lesion scrapings or biopsy, with biopsy favored when malignancy is possible. Chancroid culture can be sensitive but is limited by availability of selective media; a probable diagnosis relies on painful ulcers typical of chancroid with negative testing for syphilis and HSV. LGV diagnosis is supported by serology or by isolating C. trachomatis with confirmation of L1–L3 serovars. Molluscum can be confirmed by histology and electron microscopy. Chancres may be missed by patients, and early syphilis serology can be falsely negative; when diagnosis is uncertain or cancer is possible, biopsy is indicated.
Treatment
Medications
Chancroid can be treated with single-dose azithromycin 1 g orally or ceftriaxone 250 mg intramuscularly. Condylomata acuminata have no single best therapy; options include imiquimod 5% cream three times weekly for up to 16 weeks or podofilox 0.5% solution or gel twice daily for three days followed by one day off, repeated in cycles. Donovanosis can be treated for more than three weeks with trimethoprim-sulfamethoxazole, tetracycline, or doxycycline. Genital herpes is treated with acyclovir, famciclovir, or valacyclovir for 7–10 days, with intravenous acyclovir for severe disease. Tuberculous genital lesions require systemic antituberculosis therapy. LGV is treated with doxycycline 100 mg twice daily for 21 days. Molluscum contagiosum can be managed with lesion destruction via desiccation, cryotherapy, or curettage.
Complementary and Alternative Therapies
Alternative chancroid regimens include erythromycin for seven days or ciprofloxacin for three days. Wart management can include cryosurgery, excision, electrosurgery, or laser therapy. Donovanosis alternatives include prolonged ciprofloxacin, erythromycin, or chloramphenicol. LGV can also be treated with erythromycin four times daily for 21 days.
Ongoing Care and Follow-Up
Patients should be encouraged to have sexual partners evaluated and treated. HIV testing should be performed at diagnosis and repeated with syphilis serology about three months later if initially negative. Consider cervical cytology screening for patients evaluated for sexually transmitted infections who have not had a Pap smear within the past year, and counsel patients with HPV on the need for ongoing screening.
Pediatric Considerations
Sexually transmitted genital lesions in a child require evaluation for sexual abuse.
Complications
Untreated primary syphilis can disseminate and progress to secondary disease within weeks. Chancroid, genital herpes, and syphilis increase susceptibility to HIV transmission.
Basics
Description
Genital lesions involve the reproductive organs and can result from a range of infectious and noninfectious conditions.
Epidemiology
Genital ulcers are the most frequent sexually transmitted cause of genital lesions. In North America and Europe, the leading sexually transmitted causes of genital ulcer disease are herpes simplex and syphilis. In the United States, a substantial proportion of people aged 14–49 have HSV-2 infection, and HSV-1 accounts for a significant minority of genital herpes cases. Chancroid is more common in Africa, Asia, and Latin America but can occur in sporadic U.S. outbreaks. Lymphogranuloma venereum is uncommon in industrialized countries, though outbreaks have been reported among men who have sex with men.
Risk Factors
About half of exposed sexual partners may acquire genital warts (HPV) after contact with an infected partner. Sexual transmission of syphilis can be high, with rates reported up to roughly one-third per exposure.
General Prevention
Risk reduction includes abstinence, consistent condom use, limiting partners, safer-sex practices, and HPV vaccination. Two HPV vaccines are available for females aged 9–26 that protect against HPV 6 and 11 (responsible for most genital warts) and HPV 16 and 18 (responsible for a large proportion of cervical cancers): the quadrivalent vaccine (Gardasil) and the bivalent vaccine (Cervarix). Quadrivalent vaccination in males aged 9–26 can reduce genital warts.
Etiology
Infectious Ulcers
Common causes include genital herpes, syphilis due to Treponema pallidum, chancroid due to Haemophilus ducreyi, lymphogranuloma venereum from Chlamydia trachomatis serovars L1–L3, donovanosis (granuloma inguinale) from Klebsiella granulomatis, and less commonly tuberculosis or tularemia. Additional reported etiologies include candidiasis, histoplasmosis, amebiasis, gonorrhea, and trichomoniasis. Acute or primary HIV infection can present with genital ulcers, and a notable minority of ulcer cases involve more than one pathogen.
Noninfectious Ulcers
Noninfectious causes include trauma, fixed drug eruption, malignancy, systemic lupus erythematosus, and Behçet disease.
Other Lesion Patterns
Papules may be due to candidiasis, molluscum contagiosum, scabies, syphilis, or condylomata acuminata from HPV, with types 6 and 11 most common and types 16, 18, 31, 33, and 35 linked to cervical dysplasia. Vesicles or bullae suggest herpes, impetigo, or scabies. Diffuse erythema may occur with candidiasis, erysipelas often after trauma or surgery, contact dermatitis, drug eruption, psoriasis, or trauma. Other patterns include benign cysts, nodules from hidradenitis or furunculosis, and crusted lesions from herpes or scabies
.
Commonly Associated Conditions
HIV and other sexually transmitted infections commonly coexist with genital lesions.
Diagnosis
History
Key points include time of onset, associated symptoms, immunodeficiency, prior sexually transmitted infections, prior similar lesions, and travel. Typical incubation periods vary by cause, including herpes within days but sometimes up to weeks, genital warts usually weeks to months, syphilis typically weeks but up to months, chancroid usually within a week but variable, donovanosis over weeks, molluscum potentially months, and ectoparasites such as pubic lice or scabies often after several weeks. Lesions appearing within hours of exposure suggest trauma, chemical irritation, or hypersensitivity. Itching is common early in herpes and with scabies or pubic lice, and mild pruritus can occur in secondary syphilis.
Physical Examination
Define lesion morphology and distribution, determine whether disease is localized or generalized, assess tenderness, lymphadenopathy, and any urethral or vaginal discharge, and examine buccal mucosa and perianal skin; pelvic examination or prostate assessment is often needed. Early herpes often begins as clustered vesicles on an erythematous base, may show umbilication, is typically very painful, and is often ulcerated by presentation. Pain is typical of herpes, chancroid, and tularemia, whereas donovanosis ulcers are usually painless. Ulcer base and borders can help narrow the differential: Behçet ulcers may be yellow and necrotic, chancroid bases are often necrotic, donovanosis can appear beefy red with hypertrophy, and syphilitic and herpetic ulcers may have cleaner bases; chancroid ulcers are classically nonindurated with irregular erythematous edges, syphilitic ulcers are indurated, and herpetic ulcers often have erythematous borders. Primary syphilis more often produces a solitary lesion, while chancroid often causes multiple ulcers of varying size. Behçet disease can cause recurrent, multiple genital ulcers involving scrotum or vulva and may be accompanied by recurrent oral ulcers and skin or eye disease, which may not occur simultaneously. Linear burrows suggest scabies, and reddish specks may reflect crab louse excreta. Urethral discharge suggests gonorrhea or reactive arthritis. In adults with genital lesions, inguinal lymphadenopathy supports consideration of chancroid, LGV, syphilis, herpes, lymphoma, or tuberculosis. LGV ulcers may be unnoticed and heal spontaneously, followed weeks later by painful inguinal lymphadenopathy and systemic symptoms, and untreated disease can progress to elephantiasis-like genital distortion. Lymphadenopathy tends to be unilateral in chancroid and LGV and more often bilateral in genital herpes. Painful perianal ulcers or mucosal ulcers seen on anoscopy should be treated presumptively for HSV and LGV. Molluscum contagiosum causes mildly contagious umbilicated papules up to about 1 cm, and advanced HIV may cause widespread or large lesions. Tuberculous genital lesions may appear as chronic minimally painful red, firm, nodular sores. Clinical appearance alone is often insufficient for diagnosis.
Diagnostic Tests and Interpretation
Laboratory Studies
All patients with genital, anal, or perianal ulcers should receive syphilis serology with dark-field examination when available, HSV testing by culture or PCR or type-specific serology, and HIV testing. If suspicion and culture capacity exist, obtain H. ducreyi culture for chancroid. Donovanosis can be supported by identifying intracellular Donovan bodies on Giemsa or Wright stain from lesion scrapings or biopsy, with biopsy favored when malignancy is possible. Chancroid culture can be sensitive but is limited by availability of selective media; a probable diagnosis relies on painful ulcers typical of chancroid with negative testing for syphilis and HSV. LGV diagnosis is supported by serology or by isolating C. trachomatis with confirmation of L1–L3 serovars. Molluscum can be confirmed by histology and electron microscopy. Chancres may be missed by patients, and early syphilis serology can be falsely negative; when diagnosis is uncertain or cancer is possible, biopsy is indicated.
Treatment
Medications
Chancroid can be treated with single-dose azithromycin 1 g orally or ceftriaxone 250 mg intramuscularly. Condylomata acuminata have no single best therapy; options include imiquimod 5% cream three times weekly for up to 16 weeks or podofilox 0.5% solution or gel twice daily for three days followed by one day off, repeated in cycles. Donovanosis can be treated for more than three weeks with trimethoprim-sulfamethoxazole, tetracycline, or doxycycline. Genital herpes is treated with acyclovir, famciclovir, or valacyclovir for 7–10 days, with intravenous acyclovir for severe disease. Tuberculous genital lesions require systemic antituberculosis therapy. LGV is treated with doxycycline 100 mg twice daily for 21 days. Molluscum contagiosum can be managed with lesion destruction via desiccation, cryotherapy, or curettage.
Complementary and Alternative Therapies
Alternative chancroid regimens include erythromycin for seven days or ciprofloxacin for three days. Wart management can include cryosurgery, excision, electrosurgery, or laser therapy. Donovanosis alternatives include prolonged ciprofloxacin, erythromycin, or chloramphenicol. LGV can also be treated with erythromycin four times daily for 21 days.
Ongoing Care and Follow-Up
Patients should be encouraged to have sexual partners evaluated and treated. HIV testing should be performed at diagnosis and repeated with syphilis serology about three months later if initially negative. Consider cervical cytology screening for patients evaluated for sexually transmitted infections who have not had a Pap smear within the past year, and counsel patients with HPV on the need for ongoing screening.
Pediatric Considerations
Sexually transmitted genital lesions in a child require evaluation for sexual abuse.
Complications
Untreated primary syphilis can disseminate and progress to secondary disease within weeks. Chancroid, genital herpes, and syphilis increase susceptibility to HIV transmission.
- Published on
Infectious Diseases and Microbiology: Fever of Unknown Origin
Basics
Description
Fever of unknown origin (FUO) describes persistent fever that lasts longer than expected for a typical acute febrile illness and is meant to exclude self-limited infections, conditions identified on basic initial testing, and benign fevers below 38.3°C. Current criteria, revised from the 1961 Petersdorf and Beeson definition, require illness duration of at least three weeks, documented temperatures above 38.3°C on multiple occasions, and no diagnosis despite a thorough evaluation. Durack and Street proposed FUO subtypes reflecting differing causes and workups: classical FUO, immune-deficient FUO, healthcare-associated FUO, and HIV-related FUO. Using this framework, immune-deficient patients, hospitalized patients who were afebrile before admission, and patients with HIV may meet FUO criteria after three to five inpatient days of evaluation with 48 hours of negative cultures.
Approach to the Patient
FUO is clinically difficult for both patients and clinicians, and management centers on identifying a cause, reaching a diagnosis efficiently and safely with appropriate testing, and initiating targeted therapy once a diagnosis is established or, when necessary, performing a therapeutic trial. Evaluation should be individualized to avoid both excessive testing for unlikely causes and missed evaluation of likely ones, so rigid algorithms are discouraged. Key aims are to confirm true fever and define its pattern, assess illness tempo and severity, look for localizing symptoms, place the illness in context of comorbidities and medications, and identify exposure risks such as travel, occupational and sexual history, animal contact, vaccination status, and intravenous drug use. Minimum baseline evaluation should include detailed history, repeated physical examination, complete blood count with smear, differential and platelets, routine chemistries including liver enzymes, LDH and bilirubin, urinalysis with microscopy and culture, chest radiograph, ESR and CRP, ANA, rheumatoid factor and anti-CCP, three sets of blood cultures obtained off antibiotics, tuberculosis testing with TST or IGRA, CT abdomen and pelvis, and infectious serologies guided by the clinical scenario.
Special Populations
Geriatric Considerations
Consider polymyalgia rheumatica and recognize that the distribution of undiagnosed etiologies differs in older adults.
Pediatric Considerations
Herpesviruses HHV-6/7/8 are common in infants and children. Ultrasound is preferred over CT to limit radiation and because pediatric imaging resolution is favorable. Connective tissue disease and neoplasia are uncommon under 12 months, Kawasaki disease is common under 5 years, Still’s disease often affects children and young adults, and arthritis in children should be treated as a marker of potentially serious disease. Periodic fever and other entities to consider include hyper-IgD syndrome, cyclic neutropenia, and nephroma.
Pregnancy Considerations
Pregnancy increases venous thromboembolism risk. Consider septic pelvic thrombophlebitis after obstetric surgery, sexually transmitted infections including HIV and syphilis, and pyometra.
Epidemiology
True FUO is uncommon. Reported etiologic frequencies vary by population, geography, institution, age, local disease prevalence, and practice patterns. Over time, the proportion of infectious and malignant causes has decreased while the proportion of undiagnosed cases has increased.
Diagnosis
FUO evaluation relies on iterative history-taking, repeated examinations, laboratory studies, and imaging.
History
History should cover alcohol use, medications, occupational and sexual exposures, pets, travel, family conditions, and prior illnesses. Although fever patterns are described for many disorders, truly distinctive patterns are rare in practice; classic patterns such as malaria are uncommon in many settings, and patterns considered characteristic for other diseases, such as Pel–Ebstein fever in lymphoma, are infrequently observed.
Physical Examination
Repeated comprehensive examination is central and should specifically assess the oropharynx for dental abscess, thyroid for thyroiditis, temporal regions for arteritis, cardiac auscultation for murmurs suggesting endocarditis or atrial myxoma, skin for vasculitis and Whipple disease clues, all wounds and vascular access sites with dressings removed for inspection, regional lymph nodes, and the genitoperineal region, sometimes requiring rectal or vaginal examination. Relative bradycardia can be a clue but is nonspecific and occurs in diverse conditions including brucellosis, drug fever, factitious fever, hepatitis A, Legionella infection, leptospirosis, malignancy, psittacosis, subacute necrotizing lymphadenitis, and typhoid fever. Fever from solid tumors and many connective tissue diseases may abate with NSAIDs, whereas fever from other causes may not. Symptoms such as sweats, chills, and weight loss do not reliably distinguish etiologies. Clues suggesting factitious fever include lack of tachycardia or tachypnea, temperatures exceeding 41°C, absent diurnal variation, and lack of sweating after defervescence.
Diagnostic Tests and Interpretation
Laboratory Studies
Noninvasive tests yield a diagnosis in roughly one-quarter of cases and include serologies for infectious and rheumatologic disease, biochemical markers such as ferritin for Still’s disease, and genetic markers such as those used for familial Mediterranean fever.
Imaging
Imaging is primarily used to localize targets for further evaluation. Abdominal CT has improved diagnostic yield when followed by invasive sampling, though false negatives can occur even with solid-organ abscesses. MRI is preferred when spinal or paraspinal pathology is suspected. Nuclear imaging with gallium-67 or indium-111 labeled leukocytes can sometimes help when infection or malignancy is suspected, but limitations include false-negative gallium scans in secondarily infected lesions, poor detection of splenic abscess due to background uptake, and low positive predictive value with indium studies. FDG PET/CT can help establish malignant, inflammatory, or infectious diagnoses in at least one-third of cases. If imaging and serologies are unrevealing, invasive investigations such as liver and bone marrow biopsy should be considered, and fewer than half of diagnoses are ultimately made via biopsy or laparotomy. Temporal artery biopsy is often high yield in older adults with markedly elevated ESR even without obvious local symptoms. Disseminated tuberculosis is a highly treatable lethal cause of FUO and warrants aggressive pursuit; TST and IGRA can be negative in up to half of cases, sputum smears are only intermittently positive, and confirmation often requires biopsy of lymph node, bone marrow, or liver. Excisional lymph node biopsy is helpful when nodes are enlarged, though inguinal nodes are commonly palpable and usually not diagnostically useful.
Differential Diagnosis
The major etiologic categories and approximate frequencies include infections, malignancies, connective tissue or inflammatory disorders, miscellaneous causes, and undiagnosed cases. Unusual presentations of common diseases and conditions that are difficult to confirm account for more FUO cases than truly rare diseases, and thinking in terms of FUO subtype helps prioritize causes.
Classical FUO
Common infectious drivers remain tuberculosis, bacterial endocarditis, and intraabdominal collections. Systemic bacterial causes include bartonellosis, brucellosis, Campylobacter infection, cat-scratch disease or bacillary angiomatosis, ehrlichiosis, gonococcemia, HACEK organisms, Legionella infection, leptospirosis, listeriosis, Lyme disease, meningococcemia, rat-bite fever, relapsing fever due to Borrelia recurrentis, salmonellosis including typhoid, syphilis, tularemia, and yersiniosis, along with chlamydial, fungal, parasitic, and viral infections. Localized infections include intravascular infections such as endocarditis, aortitis, catheter infections, septic jugular phlebitis, and vascular graft infection; intraabdominal infections such as appendicitis, cholangitis, cholecystitis, diverticulitis, and diverse abscesses (subphrenic, hepatic, splenic, pancreatic, perinephric, pelvic), mesenteric lymphadenitis, pelvic inflammatory disease, and pyometra; prosthesis-related infections involving joint prostheses, pacemakers, defibrillators, shunts, and vascular access; and other focal infections including dental, intracranial, pulmonary, mastoid, middle ear, sinus, prostatic, and wound infections. Common noninfectious causes include malignancies such as renal cell carcinoma, hepatocellular carcinoma, Hodgkin and non-Hodgkin lymphoma, colon cancer, leukemia, malignant histiocytosis, pancreatic cancer, and sarcoma; benign tumors such as atrial myxoma; inflammatory disorders such as adult Still’s disease, Behçet syndrome, and cryoglobulinemia; and miscellaneous conditions including neuroleptic malignant syndrome, hematoma, recurrent pulmonary embolism, aortic dissection, post–myocardial infarction fever syndromes, subacute thyroiditis, hyperthyroidism, adrenal insufficiency, drug fever, factitious fever, gout, pseudogout, hypersensitivity pneumonitis, stroke, familial Mediterranean fever, and pheochromocytoma.
Healthcare-Associated FUO
Infectious causes include intravascular catheter infection, septic thrombophlebitis, prosthetic device infection, deep postoperative wound infections and intraabdominal collections, C. difficile colitis that may present with ileus rather than diarrhea in severe disease, septic pelvic thrombophlebitis after obstetric or gynecologic surgery, acalculous cholecystitis, and ICU-associated sinusitis related to nasogastric tubes and endotracheal intubation. Miscellaneous causes include venous thromboembolism, drug fever often without rash or eosinophilia and commonly triggered by antimicrobials and multiple other medication classes, factitious fever, and postoperative inflammatory fever without a defined diagnosis.
Immune-Deficient FUO
Most cases are infectious, with pathogen identification in a large fraction, and the likely infections depend on the type and duration of immunodeficiency. Persistent fever can reflect progressive or relapsed malignancy in hematology-oncology patients, inflammatory causes such as graft-versus-host disease, or miscellaneous etiologies including venous thromboembolism, drug fever, and adrenal insufficiency.
HIV-Related FUO
Most cases are infectious, though FUO has decreased in the antiretroviral era due to fewer opportunistic infections, and likely etiologies depend strongly on CD4 count. Scenarios include HIV seroconversion illness, opportunistic infections such as mycobacterial disease, Pneumocystis jirovecii, cytomegalovirus, toxoplasmosis, histoplasmosis, and leishmaniasis, inflammatory syndromes such as immune reconstitution, Castleman disease, and hemophagocytic lymphohistiocytosis, malignancies such as lymphoma and Kaposi sarcoma, and drug fever.
Treatment
Medications
Management is determined by the underlying cause. If TST or IGRA is positive or if granulomatous disease with possible anergy is suspected, a therapeutic trial for tuberculosis is recommended and may be continued for up to six weeks; persistent fever beyond this period suggests an alternate diagnosis. Glucocorticoids and NSAIDs can suppress fever and obscure diagnosis while allowing infection to progress and should be avoided unless infection has largely been excluded. In severely immune-deficient patients, empiric antimicrobial therapy is often necessary because infections are common and associated with high mortality.
Ongoing Care and Follow-Up
Hospitalized patients with FUO require close monitoring for evolving symptoms or new physical findings that may provide diagnostic direction.
Basics
Description
Fever of unknown origin (FUO) describes persistent fever that lasts longer than expected for a typical acute febrile illness and is meant to exclude self-limited infections, conditions identified on basic initial testing, and benign fevers below 38.3°C. Current criteria, revised from the 1961 Petersdorf and Beeson definition, require illness duration of at least three weeks, documented temperatures above 38.3°C on multiple occasions, and no diagnosis despite a thorough evaluation. Durack and Street proposed FUO subtypes reflecting differing causes and workups: classical FUO, immune-deficient FUO, healthcare-associated FUO, and HIV-related FUO. Using this framework, immune-deficient patients, hospitalized patients who were afebrile before admission, and patients with HIV may meet FUO criteria after three to five inpatient days of evaluation with 48 hours of negative cultures.
Approach to the Patient
FUO is clinically difficult for both patients and clinicians, and management centers on identifying a cause, reaching a diagnosis efficiently and safely with appropriate testing, and initiating targeted therapy once a diagnosis is established or, when necessary, performing a therapeutic trial. Evaluation should be individualized to avoid both excessive testing for unlikely causes and missed evaluation of likely ones, so rigid algorithms are discouraged. Key aims are to confirm true fever and define its pattern, assess illness tempo and severity, look for localizing symptoms, place the illness in context of comorbidities and medications, and identify exposure risks such as travel, occupational and sexual history, animal contact, vaccination status, and intravenous drug use. Minimum baseline evaluation should include detailed history, repeated physical examination, complete blood count with smear, differential and platelets, routine chemistries including liver enzymes, LDH and bilirubin, urinalysis with microscopy and culture, chest radiograph, ESR and CRP, ANA, rheumatoid factor and anti-CCP, three sets of blood cultures obtained off antibiotics, tuberculosis testing with TST or IGRA, CT abdomen and pelvis, and infectious serologies guided by the clinical scenario.
Special Populations
Geriatric Considerations
Consider polymyalgia rheumatica and recognize that the distribution of undiagnosed etiologies differs in older adults.
Pediatric Considerations
Herpesviruses HHV-6/7/8 are common in infants and children. Ultrasound is preferred over CT to limit radiation and because pediatric imaging resolution is favorable. Connective tissue disease and neoplasia are uncommon under 12 months, Kawasaki disease is common under 5 years, Still’s disease often affects children and young adults, and arthritis in children should be treated as a marker of potentially serious disease. Periodic fever and other entities to consider include hyper-IgD syndrome, cyclic neutropenia, and nephroma.
Pregnancy Considerations
Pregnancy increases venous thromboembolism risk. Consider septic pelvic thrombophlebitis after obstetric surgery, sexually transmitted infections including HIV and syphilis, and pyometra.
Epidemiology
True FUO is uncommon. Reported etiologic frequencies vary by population, geography, institution, age, local disease prevalence, and practice patterns. Over time, the proportion of infectious and malignant causes has decreased while the proportion of undiagnosed cases has increased.
Diagnosis
FUO evaluation relies on iterative history-taking, repeated examinations, laboratory studies, and imaging.
History
History should cover alcohol use, medications, occupational and sexual exposures, pets, travel, family conditions, and prior illnesses. Although fever patterns are described for many disorders, truly distinctive patterns are rare in practice; classic patterns such as malaria are uncommon in many settings, and patterns considered characteristic for other diseases, such as Pel–Ebstein fever in lymphoma, are infrequently observed.
Physical Examination
Repeated comprehensive examination is central and should specifically assess the oropharynx for dental abscess, thyroid for thyroiditis, temporal regions for arteritis, cardiac auscultation for murmurs suggesting endocarditis or atrial myxoma, skin for vasculitis and Whipple disease clues, all wounds and vascular access sites with dressings removed for inspection, regional lymph nodes, and the genitoperineal region, sometimes requiring rectal or vaginal examination. Relative bradycardia can be a clue but is nonspecific and occurs in diverse conditions including brucellosis, drug fever, factitious fever, hepatitis A, Legionella infection, leptospirosis, malignancy, psittacosis, subacute necrotizing lymphadenitis, and typhoid fever. Fever from solid tumors and many connective tissue diseases may abate with NSAIDs, whereas fever from other causes may not. Symptoms such as sweats, chills, and weight loss do not reliably distinguish etiologies. Clues suggesting factitious fever include lack of tachycardia or tachypnea, temperatures exceeding 41°C, absent diurnal variation, and lack of sweating after defervescence.
Diagnostic Tests and Interpretation
Laboratory Studies
Noninvasive tests yield a diagnosis in roughly one-quarter of cases and include serologies for infectious and rheumatologic disease, biochemical markers such as ferritin for Still’s disease, and genetic markers such as those used for familial Mediterranean fever.
Imaging
Imaging is primarily used to localize targets for further evaluation. Abdominal CT has improved diagnostic yield when followed by invasive sampling, though false negatives can occur even with solid-organ abscesses. MRI is preferred when spinal or paraspinal pathology is suspected. Nuclear imaging with gallium-67 or indium-111 labeled leukocytes can sometimes help when infection or malignancy is suspected, but limitations include false-negative gallium scans in secondarily infected lesions, poor detection of splenic abscess due to background uptake, and low positive predictive value with indium studies. FDG PET/CT can help establish malignant, inflammatory, or infectious diagnoses in at least one-third of cases. If imaging and serologies are unrevealing, invasive investigations such as liver and bone marrow biopsy should be considered, and fewer than half of diagnoses are ultimately made via biopsy or laparotomy. Temporal artery biopsy is often high yield in older adults with markedly elevated ESR even without obvious local symptoms. Disseminated tuberculosis is a highly treatable lethal cause of FUO and warrants aggressive pursuit; TST and IGRA can be negative in up to half of cases, sputum smears are only intermittently positive, and confirmation often requires biopsy of lymph node, bone marrow, or liver. Excisional lymph node biopsy is helpful when nodes are enlarged, though inguinal nodes are commonly palpable and usually not diagnostically useful.
Differential Diagnosis
The major etiologic categories and approximate frequencies include infections, malignancies, connective tissue or inflammatory disorders, miscellaneous causes, and undiagnosed cases. Unusual presentations of common diseases and conditions that are difficult to confirm account for more FUO cases than truly rare diseases, and thinking in terms of FUO subtype helps prioritize causes.
Classical FUO
Common infectious drivers remain tuberculosis, bacterial endocarditis, and intraabdominal collections. Systemic bacterial causes include bartonellosis, brucellosis, Campylobacter infection, cat-scratch disease or bacillary angiomatosis, ehrlichiosis, gonococcemia, HACEK organisms, Legionella infection, leptospirosis, listeriosis, Lyme disease, meningococcemia, rat-bite fever, relapsing fever due to Borrelia recurrentis, salmonellosis including typhoid, syphilis, tularemia, and yersiniosis, along with chlamydial, fungal, parasitic, and viral infections. Localized infections include intravascular infections such as endocarditis, aortitis, catheter infections, septic jugular phlebitis, and vascular graft infection; intraabdominal infections such as appendicitis, cholangitis, cholecystitis, diverticulitis, and diverse abscesses (subphrenic, hepatic, splenic, pancreatic, perinephric, pelvic), mesenteric lymphadenitis, pelvic inflammatory disease, and pyometra; prosthesis-related infections involving joint prostheses, pacemakers, defibrillators, shunts, and vascular access; and other focal infections including dental, intracranial, pulmonary, mastoid, middle ear, sinus, prostatic, and wound infections. Common noninfectious causes include malignancies such as renal cell carcinoma, hepatocellular carcinoma, Hodgkin and non-Hodgkin lymphoma, colon cancer, leukemia, malignant histiocytosis, pancreatic cancer, and sarcoma; benign tumors such as atrial myxoma; inflammatory disorders such as adult Still’s disease, Behçet syndrome, and cryoglobulinemia; and miscellaneous conditions including neuroleptic malignant syndrome, hematoma, recurrent pulmonary embolism, aortic dissection, post–myocardial infarction fever syndromes, subacute thyroiditis, hyperthyroidism, adrenal insufficiency, drug fever, factitious fever, gout, pseudogout, hypersensitivity pneumonitis, stroke, familial Mediterranean fever, and pheochromocytoma.
Healthcare-Associated FUO
Infectious causes include intravascular catheter infection, septic thrombophlebitis, prosthetic device infection, deep postoperative wound infections and intraabdominal collections, C. difficile colitis that may present with ileus rather than diarrhea in severe disease, septic pelvic thrombophlebitis after obstetric or gynecologic surgery, acalculous cholecystitis, and ICU-associated sinusitis related to nasogastric tubes and endotracheal intubation. Miscellaneous causes include venous thromboembolism, drug fever often without rash or eosinophilia and commonly triggered by antimicrobials and multiple other medication classes, factitious fever, and postoperative inflammatory fever without a defined diagnosis.
Immune-Deficient FUO
Most cases are infectious, with pathogen identification in a large fraction, and the likely infections depend on the type and duration of immunodeficiency. Persistent fever can reflect progressive or relapsed malignancy in hematology-oncology patients, inflammatory causes such as graft-versus-host disease, or miscellaneous etiologies including venous thromboembolism, drug fever, and adrenal insufficiency.
HIV-Related FUO
Most cases are infectious, though FUO has decreased in the antiretroviral era due to fewer opportunistic infections, and likely etiologies depend strongly on CD4 count. Scenarios include HIV seroconversion illness, opportunistic infections such as mycobacterial disease, Pneumocystis jirovecii, cytomegalovirus, toxoplasmosis, histoplasmosis, and leishmaniasis, inflammatory syndromes such as immune reconstitution, Castleman disease, and hemophagocytic lymphohistiocytosis, malignancies such as lymphoma and Kaposi sarcoma, and drug fever.
Treatment
Medications
Management is determined by the underlying cause. If TST or IGRA is positive or if granulomatous disease with possible anergy is suspected, a therapeutic trial for tuberculosis is recommended and may be continued for up to six weeks; persistent fever beyond this period suggests an alternate diagnosis. Glucocorticoids and NSAIDs can suppress fever and obscure diagnosis while allowing infection to progress and should be avoided unless infection has largely been excluded. In severely immune-deficient patients, empiric antimicrobial therapy is often necessary because infections are common and associated with high mortality.
Ongoing Care and Follow-Up
Hospitalized patients with FUO require close monitoring for evolving symptoms or new physical findings that may provide diagnostic direction.

