- Published on
Acquired Immunodeficiency Syndrome (AIDS)
Acquired immunodeficiency syndrome (AIDS) is the advanced stage of infection caused by the human immunodeficiency virus (HIV). It is characterized by progressive destruction of cell-mediated immunity, particularly CD4⁺ T lymphocytes, which leads to opportunistic infections, malignancies, and systemic complications. A diagnosis of AIDS is made in a person with confirmed HIV infection when the CD4 cell count falls below 200 cells/mm³ (or <14% of total lymphocytes) or when specific aids-defining conditions are present, even if symptoms otherwise minimal.< />pan>
In the United States, approximately 1.2 million people are living with HIV, and a subset have progressed to AIDS. New diagnoses occur disproportionately among Black/African American and Latino/Hispanic populations, and the majority of cases are among men, particularly gay, bisexual, and other men who have sex with men (MSM). The most commonly affected age group is 25 to 59 years. Although there is no proven hereditary predisposition, individuals with certain genetic mutations, such as CCR5 deletions, are less susceptible to HIV infection and may progress more slowly to AIDS. Without treatment, HIV can also be transmitted vertically from mother to fetus or neonate.
Clinical presentation varies widely. Some individuals are asymptomatic, while others present with nonspecific symptoms such as fever, weight loss, anorexia, and fatigue. As immune function declines, patients develop opportunistic infections involving the lungs (e.g., Pneumocystis jirovecii pneumonia, tuberculosis), central nervous system (e.g., toxoplasmosis, cryptococcosis, progressive multifocal leukoencephalopathy), gastrointestinal tract (e.g., chronic cryptosporidiosis, CMV colitis), eyes (e.g., CMV retinitis), and skin. AIDS-related malignancies include Kaposi sarcoma, primary CNS lymphoma, invasive cervical cancer, and high-grade B-cell non-Hodgkin lymphomas.
HIV infection is caused by HIV-1 (most common worldwide) or HIV-2 (less common, mainly in West Africa). Transmission occurs through sexual contact, needle sharing during injection drug use, transfusion of contaminated blood products, and vertical transmission from mother to child during pregnancy, delivery, or breastfeeding.
Diagnosis relies on HIV antibody/antigen testing, followed by assessment of immune and virologic status. Key laboratory studies include CD4 cell count to gauge immunodeficiency and HIV viral load (HIV RNA PCR) to guide therapy and monitor treatment response. Additional evaluation commonly includes screening for sexually transmitted infections, hepatitis A, B, and C, syphilis, toxoplasmosis, and tuberculosis. Imaging studies such as chest radiography or CT and brain MRI or CT are used when pulmonary or neurologic symptoms suggest opportunistic disease.
The cornerstone of management is early and sustained antiretroviral therapy (ART), which should be initiated in all patients with HIV regardless of CD4 count. ART suppresses viral replication, allows immune recovery, reduces opportunistic infections, and dramatically improves survival. Modern regimens typically combine two nucleoside reverse transcriptase inhibitors with an integrase inhibitor, and are generally well tolerated. In addition to ART, patients with AIDS often require prophylactic therapy to prevent infections such as Pneumocystis jirovecii pneumonia and Mycobacterium avium complex until immune reconstitution occurs.
Nonpharmacologic management includes maintaining adequate nutrition, practicing safe sex, avoiding needle sharing, and minimizing exposure to opportunistic pathogens. Vaccinations against influenza, pneumococcus, hepatitis A and B, meningococcus, COVID-19, and Tdap are recommended, while live vaccines are generally avoided in patients with advanced immunosuppression.
The prognosis of HIV/AIDS has changed dramatically. With consistent access to ART and expert medical follow-up, HIV is now a chronic, manageable condition, and many patients enjoy near-normal life expectancy and quality of life. All individuals diagnosed with HIV or AIDS should be referred to clinicians experienced in HIV care to ensure optimal treatment, monitoring, and prevention of complications.
- Published on
KembaraXtra-Medicine- Acromegaly
Acromegaly is a chronic, progressive disorder caused by excess secretion of growth hormone (GH), which in turn stimulates increased production of insulin-like growth factor 1 (IGF-1). The disease develops insidiously, often over many years, and may lead to significant morbidity and mortality if left untreated, particularly from cardiovascular and respiratory complications.
The ICD-10-CM code for acromegaly and pituitary gigantism is E22.0. The incidence is approximately 3 to 4 new cases per 1 million people annually, with a prevalence of 50 to 60 cases per 1 million, and some estimates as high as 90 per 1 million. Acromegaly occurs slightly more often in women. The typical age at diagnosis is around 40 years in men and 45 years in women.
Clinically, patients develop coarse facial features due to progressive soft tissue overgrowth. Common findings include coarse, oily skin, excessive sweating, and macrocephaly. Enlargement of the hands and feet produces broad, spade-like, fleshy extremities, often accompanied by a history of increasing ring, glove, hat, or shoe size. Facial changes such as enlargement of the nose, brow, and jaw, along with prognathism, may result in an underbite.
Musculoskeletal symptoms are common and include muscle weakness, decreased exercise tolerance, arthralgias, severe osteoarthritis, and vertebral fractures. Neurologic and compressive symptoms from the pituitary tumor may include headaches and visual field defects. Carpal tunnel syndrome is frequently seen due to soft tissue swelling.
Acromegaly is also associated with multiple systemic complications. These include diabetes mellitus, hypertension, mild to moderate obesity, and severe obstructive sleep apnea, often related to macroglossia and upper airway soft tissue enlargement. Cardiac involvement is a major contributor to morbidity and mortality and may manifest as cardiomyopathy, left ventricular hypertrophy, valvular heart disease, diastolic dysfunction, and arrhythmias. Patients may also develop benign tumors such as skin tags, uterine fibroids, prostate hypertrophy, and colon polyps, and women may experience menstrual dysfunction.
In most cases, acromegaly results from a pituitary adenoma arising from the anterior pituitary gland. Rarely, it is caused by ectopic secretion of GH or growth hormone–releasing hormone (GHRH) from nonpituitary tumors. Conditions that may mimic or be associated with acromegaly include neuroendocrine tumors, McCune-Albright syndrome, and pachydermoperiostosis.
Diagnosis begins with biochemical testing followed by imaging to localize the source of hormone excess. The initial screening test is a serum IGF-1 level; a normal IGF-1 level makes acromegaly unlikely. If IGF-1 is elevated or equivocal, an oral glucose tolerance test is performed. Failure of GH to suppress to below 1 ng/mL after a glucose load confirms the diagnosis. Random GH measurements are not recommended because of the hormone’s pulsatile secretion.
Once biochemical confirmation is obtained, imaging is used to identify the tumor. MRI of the pituitary and hypothalamus is the preferred modality, while CT scanning is reserved for patients who cannot undergo MRI.
The treatment of choice is transsphenoidal microsurgical removal of the pituitary adenoma. Surgical success depends on tumor size, with failure rates of approximately 10% to 20% for microadenomas and 25% to 50% for macroadenomas confined to the sella. When surgery is contraindicated, incomplete, or when rapid control is needed before surgery (such as in patients with severe sleep apnea or heart failure), medical therapy is indicated.
Medical treatment options include somatostatin receptor ligands (octreotide, lanreotide, pasireotide), which suppress GH secretion and are commonly used preoperatively or postoperatively in patients with residual disease. Pegvisomant, a GH receptor antagonist, is particularly effective for moderate to severe persistent disease or in patients intolerant of somatostatin analogs. Combination therapy with somatostatin analogs and pegvisomant can achieve very high biochemical control rates. Cabergoline, a dopamine agonist, may be used in patients with mild residual disease or as adjunct therapy.
Radiotherapy, including stereotactic radiosurgery (gamma knife), is reserved for patients with persistent or recurrent disease after surgery who do not respond adequately to medical therapy. A major long-term complication of radiotherapy is hypopituitarism, which can occur in up to 50% of patients, necessitating lifelong endocrine follow-up.
If left untreated, acromegaly is associated with an increased mortality rate, mainly due to cardiovascular and respiratory complications. Achieving biochemical control significantly reduces the risk of diabetes and cardiovascular disease. While treatment can reverse many soft tissue and metabolic changes, bone and joint abnormalities are generally irreversible, highlighting the importance of early diagnosis and management.
A key clinical pearl is that GH excess occurring before epiphyseal closure in children and adolescents results in pituitary gigantism, rather than acromegaly. The safety and efficacy of somatostatin receptor ligands and pegvisomant during pregnancy remain uncertain.

- Published on
KembaraXtra-Medicine- Acromioclavicular Dislocation
The acromioclavicular (AC) joint is formed by the articulation between the distal clavicle and the acromion and is located on the anterosuperior aspect of the shoulder. This joint is stabilized primarily by the acromioclavicular (AC) ligaments and the coracoclavicular (CC) ligaments. An acromioclavicular dislocation, also referred to as an AC joint separation, occurs when trauma disrupts these stabilizing structures. The severity of injury can range from a mild ligament sprain to complete rupture of the AC and CC ligaments, with the degree of damage correlating directly with the amount of force applied to the shoulder.
AC joint dislocations most commonly result from direct trauma to the shoulder, such as a blow that drives the acromion inferiorly relative to the clavicle. These injuries are frequently seen in contact sports like football, rugby, lacrosse, and ice hockey, as well as in high-impact motor vehicle accidents. Falls, including falls onto an outstretched hand, can also produce AC joint injuries, though high-energy mechanisms are more likely to result in severe dislocations.
The incidence of AC joint dislocations in the general population ranges from 0.8 to 4.6 per 10,000 people per year, accounting for approximately 10% of shoulder girdle injuries in urban populations. Peak incidence occurs in individuals aged 29 to 40 years, and these injuries are most commonly seen in patients younger than 40 years. Males are more frequently affected than females. No genetic predisposition has been identified.
Clinically, patients typically present with anterior-superior shoulder pain following trauma. Physical examination may reveal visible deformity when compared with the contralateral shoulder, particularly in higher-grade injuries. A classic finding is the “piano key sign,” in which the distal clavicle appears elevated and rebounds when downward pressure is applied. Patients may also have localized tenderness, swelling, and limited shoulder motion. It is essential to assess neurovascular status, evaluate clavicular mobility, and examine surrounding joints and soft tissues for associated injuries.
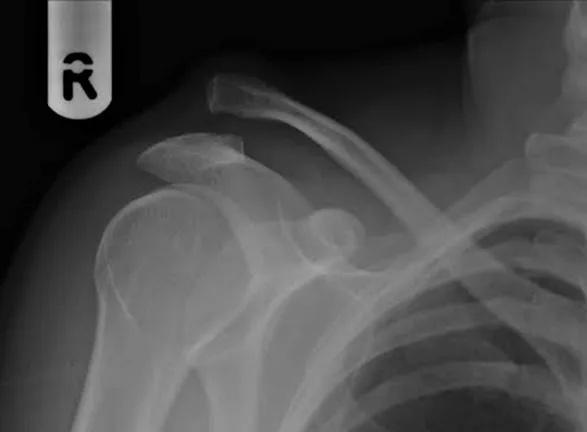
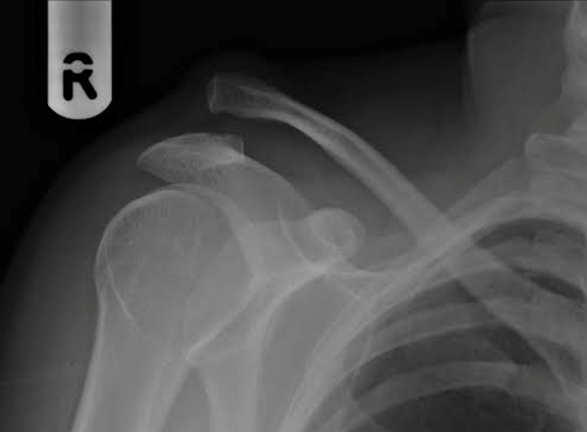
Diagnosis is often made based on clinical presentation and plain radiographs. Standard imaging includes bilateral anteroposterior views of the AC joints, axillary lateral views, Zanca views, and cross-body adduction views. Upright imaging is preferred, as supine films may underestimate injury severity. The degree of displacement is classified using the Rockwood classification, which categorizes injuries from Type I (ligament sprain) to Type VI (severe inferior displacement of the clavicle). This classification system guides treatment decisions.
Management of AC joint dislocations depends largely on injury severity. Type I and Type II injuries are treated nonoperatively with rest, ice, NSAIDs, sling immobilization, activity modification, and physical therapy. Type III injuries remain controversial; many patients achieve good outcomes with conservative management, though orthopedic consultation is recommended. Surgical intervention may be considered in patients with persistent symptoms despite nonoperative treatment. Type IV, V, and VI injuries typically require surgical management and should be referred promptly to an orthopedic surgeon.
Nonoperative treatment focuses on pain control, immobilization, and gradual rehabilitation to restore range of motion and strength. Operative treatment usually involves open reduction with reconstruction of the coracoclavicular ligaments and repair of the deltotrapezial fascia. There is no single superior surgical technique, and outcomes are generally comparable across methods. Chronic injuries may be managed either conservatively or surgically depending on symptoms and functional impairment.
Most patients managed nonoperatively recover well, regaining near-normal function by 6 weeks and returning to full activity by approximately 12 weeks. Surgical cases require a longer recovery period, often involving 6 weeks of immobilization and return to full activity by 6 months. While many patients are left with a visible deformity at the AC joint, it is often cosmetic and asymptomatic.
Preventive strategies include the use of proper shoulder protective padding in contact sports and education on safe falling techniques in activities with a high risk of falls. Patients and families should be counseled on both nonoperative and operative treatment options, expected recovery timelines, and the possibility of persistent but painless deformity following healing.
- Published on
KembaraXtra-Medicine- Actinomycosis
Actinomycosis is an indolent, slowly progressive bacterial infection caused by anaerobic or microaerophilic organisms of the genus Actinomyces. These bacteria normally colonize the mouth, gastrointestinal tract, and female genital tract and do not exist freely in the environment. Disease occurs only when there is disruption of normal mucosal barriers, allowing organisms to invade deeper tissues. Actinomycosis is characterized by painful abscess formation, dense soft tissue infiltration, and the development of draining sinus tracts.
Actinomycosis occurs worldwide but has become relatively rare, with an estimated incidence far lower than the historical rate of approximately 1 in 300,000 reported in the 1970s. Improved oral hygiene and widespread antibiotic use have contributed to this decline. Males are affected more frequently than females, with a ratio of approximately 3:1, and the disease can occur at any age, though it is most commonly seen in middle-aged adults.
Clinically, actinomycosis can involve almost any organ system, most often following local tissue injury or mucosal breach. The most common form is cervicofacial actinomycosis, accounting for about 60% of cases. This form typically arises in the setting of poor dental hygiene, recent dental procedures, or minor oral trauma. Patients may present with painful soft tissue swelling near the angle of the mandible, fever, weight loss, trismus, and draining sinus tracts or fistulas.
Thoracic actinomycosis usually results from aspiration of oral secretions containing Actinomyces species, particularly in patients with poor oral hygiene. Symptoms include fever, cough, weight loss, and pleuritic chest pain. The infection may extend beyond the lungs to involve the pleura, mediastinum, or chest wall, leading to complications such as empyema, pericarditis, chest wall sinus tracts, or tracheoesophageal fistulas.
Abdominal actinomycosis commonly follows gastrointestinal surgery, appendicitis, bowel perforation, or diverticulitis. It most often affects the ileocecal region and presents with abdominal pain, fever, weight loss, and sometimes a palpable mass. Extension to adjacent organs such as the liver can result in abscess formation and jaundice, and sinus tracts may extend to the abdominal wall. Pelvic actinomycosis may occur by contiguous spread, particularly in women, and has been associated with intrauterine device (IUD) use.
Actinomycosis is most frequently caused by Actinomyces israelii, though other species such as A. naeslundii, A. odontolyticus, A. viscosus, and A. meyeri may be involved. These organisms are gram-positive, filamentous, non–spore-forming rods. Infections are typically polymicrobial, often involving organisms such as Streptococcus, Eikenella corrodens, Bacteroides, Enterococcus, and Fusobacterium species. Predisposing factors include diabetes mellitus, malnutrition, immunosuppression, and chronic mucosal injury.
Diagnosis relies on isolating Actinomyces organisms in the appropriate clinical context. Specimens are obtained through abscess aspiration, sinus tract excision, or tissue biopsy and must be cultured under anaerobic conditions for at least 5 to 7 days, as growth is slow. A characteristic diagnostic feature is the presence of sulfur granules in pus or tissue specimens. These yellow granules represent dense colonies of Actinomyces and are diagnostic when identified microscopically, although they may take weeks to grow in culture.
Imaging studies are useful adjuncts to define the extent of disease and localize infection. Chest radiography and computed tomography (CT) scans of the head, chest, abdomen, or pelvis are commonly used depending on the suspected site of involvement. Imaging often reveals infiltrative masses that cross tissue planes, a hallmark of actinomycosis.
Treatment of actinomycosis requires prolonged antibiotic therapy and, in many cases, surgical intervention. Nonpharmacologic management includes incision and drainage of abscesses and excision of sinus tracts when indicated. Penicillin remains the treatment of choice. Initial therapy typically consists of intravenous penicillin or ampicillin for 4 to 6 weeks, followed by prolonged oral therapy with penicillin V for an additional 4 to 6 weeks or longer. In complicated cases, total treatment duration may extend to 6 to 12 months. For penicillin-allergic patients, alternatives include doxycycline, erythromycin, or clindamycin.
With appropriate treatment, the prognosis of actinomycosis is excellent. Untreated disease, however, may spread extensively to contiguous tissues while ignoring normal anatomic boundaries. Hematogenous spread is rare. Because relapse can occur if therapy is too short, long-term antibiotic treatment is essential. Referral to an infectious disease specialist is recommended when actinomycosis is suspected, and surgical consultation is often necessary for management of abscesses, sinus tracts, or necrotic tissue.
Important clinical considerations include the absence of person-to-person transmission and the fact that isolation of Actinomyces in an asymptomatic individual does not indicate disease. Pelvic actinomycosis should be considered in women with long-term IUD use, and central nervous system involvement, though uncommon, can occur in the form of brain abscesses.
Actinomycosis is an indolent, slowly progressive bacterial infection caused by anaerobic or microaerophilic organisms of the genus Actinomyces. These bacteria normally colonize the mouth, gastrointestinal tract, and female genital tract and do not exist freely in the environment. Disease occurs only when there is disruption of normal mucosal barriers, allowing organisms to invade deeper tissues. Actinomycosis is characterized by painful abscess formation, dense soft tissue infiltration, and the development of draining sinus tracts.
Actinomycosis occurs worldwide but has become relatively rare, with an estimated incidence far lower than the historical rate of approximately 1 in 300,000 reported in the 1970s. Improved oral hygiene and widespread antibiotic use have contributed to this decline. Males are affected more frequently than females, with a ratio of approximately 3:1, and the disease can occur at any age, though it is most commonly seen in middle-aged adults.
Clinically, actinomycosis can involve almost any organ system, most often following local tissue injury or mucosal breach. The most common form is cervicofacial actinomycosis, accounting for about 60% of cases. This form typically arises in the setting of poor dental hygiene, recent dental procedures, or minor oral trauma. Patients may present with painful soft tissue swelling near the angle of the mandible, fever, weight loss, trismus, and draining sinus tracts or fistulas.
Thoracic actinomycosis usually results from aspiration of oral secretions containing Actinomyces species, particularly in patients with poor oral hygiene. Symptoms include fever, cough, weight loss, and pleuritic chest pain. The infection may extend beyond the lungs to involve the pleura, mediastinum, or chest wall, leading to complications such as empyema, pericarditis, chest wall sinus tracts, or tracheoesophageal fistulas.
Abdominal actinomycosis commonly follows gastrointestinal surgery, appendicitis, bowel perforation, or diverticulitis. It most often affects the ileocecal region and presents with abdominal pain, fever, weight loss, and sometimes a palpable mass. Extension to adjacent organs such as the liver can result in abscess formation and jaundice, and sinus tracts may extend to the abdominal wall. Pelvic actinomycosis may occur by contiguous spread, particularly in women, and has been associated with intrauterine device (IUD) use.
Actinomycosis is most frequently caused by Actinomyces israelii, though other species such as A. naeslundii, A. odontolyticus, A. viscosus, and A. meyeri may be involved. These organisms are gram-positive, filamentous, non–spore-forming rods. Infections are typically polymicrobial, often involving organisms such as Streptococcus, Eikenella corrodens, Bacteroides, Enterococcus, and Fusobacterium species. Predisposing factors include diabetes mellitus, malnutrition, immunosuppression, and chronic mucosal injury.
Diagnosis relies on isolating Actinomyces organisms in the appropriate clinical context. Specimens are obtained through abscess aspiration, sinus tract excision, or tissue biopsy and must be cultured under anaerobic conditions for at least 5 to 7 days, as growth is slow. A characteristic diagnostic feature is the presence of sulfur granules in pus or tissue specimens. These yellow granules represent dense colonies of Actinomyces and are diagnostic when identified microscopically, although they may take weeks to grow in culture.
Imaging studies are useful adjuncts to define the extent of disease and localize infection. Chest radiography and computed tomography (CT) scans of the head, chest, abdomen, or pelvis are commonly used depending on the suspected site of involvement. Imaging often reveals infiltrative masses that cross tissue planes, a hallmark of actinomycosis.
Treatment of actinomycosis requires prolonged antibiotic therapy and, in many cases, surgical intervention. Nonpharmacologic management includes incision and drainage of abscesses and excision of sinus tracts when indicated. Penicillin remains the treatment of choice. Initial therapy typically consists of intravenous penicillin or ampicillin for 4 to 6 weeks, followed by prolonged oral therapy with penicillin V for an additional 4 to 6 weeks or longer. In complicated cases, total treatment duration may extend to 6 to 12 months. For penicillin-allergic patients, alternatives include doxycycline, erythromycin, or clindamycin.
With appropriate treatment, the prognosis of actinomycosis is excellent. Untreated disease, however, may spread extensively to contiguous tissues while ignoring normal anatomic boundaries. Hematogenous spread is rare. Because relapse can occur if therapy is too short, long-term antibiotic treatment is essential. Referral to an infectious disease specialist is recommended when actinomycosis is suspected, and surgical consultation is often necessary for management of abscesses, sinus tracts, or necrotic tissue.
Important clinical considerations include the absence of person-to-person transmission and the fact that isolation of Actinomyces in an asymptomatic individual does not indicate disease. Pelvic actinomycosis should be considered in women with long-term IUD use, and central nervous system involvement, though uncommon, can occur in the form of brain abscesses.
- Published on
KembaraXtra-Medicine- Actinic Keratosis
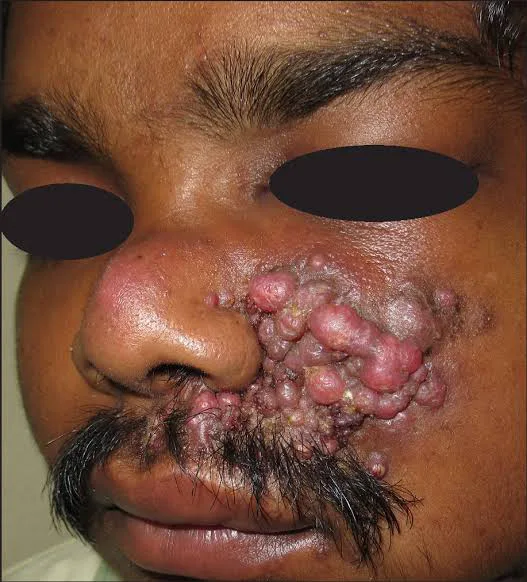
Actinic keratoses (AKs) are common premalignant skin lesions that typically present as multiple, ill-defined pink to yellow-brown, dry, scaly patches or plaques. They occur predominantly on chronically sun-exposed areas and are most often seen in middle-aged and elderly individuals. AKs are also known as solar keratoses or senile keratoses and are classified under ICD-10-CM code L57.0.
AKs are highly prevalent worldwide, with frequency varying by geographic location and sun exposure. In the northern hemisphere, approximately 11% to 25% of adults have at least one AK, whereas in regions closer to the equator, prevalence rises to 40% to 60%. In the United States, tens of millions of cases are estimated annually. The condition is more common in fair-skinned individuals, particularly those who burn rather than tan, and prevalence increases significantly with age. Men are affected more often than women, especially between the ages of 65 and 74 years.
Clinically, AKs appear on sun-damaged skin, most commonly involving the face, neck, scalp, ears, dorsal hands, and forearms. Early lesions may present as subtle erythematous patches with minimal scaling, while more advanced lesions develop thicker, yellowish, hyperkeratotic scales and feel rough or gritty on palpation. Lesions are usually less than 1 cm in diameter. Surrounding skin often shows signs of chronic photodamage, including atrophy, pigmentary changes, and telangiectasia. Several clinical variants exist, such as hypertrophic AKs with cutaneous horns, lichenoid AKs, proliferative AKs, pigmented AKs, and actinic cheilitis involving the lips.
The primary etiologic factor in the development of AKs is chronic ultraviolet (UV) radiation exposure. Additional contributing factors include ionizing radiation, arsenic exposure, polycyclic hydrocarbons, immunosuppression, human papillomavirus infection, cigarette smoke, and chronic skin injury. Genetic susceptibility also plays a role, particularly in conditions such as xeroderma pigmentosum and other inherited disorders associated with impaired DNA repair or increased skin cancer risk. Mutations in the p53 tumor suppressor gene are commonly implicated.
Diagnosis of actinic keratosis is usually clinical, based on characteristic appearance and patient history of sun exposure. A biopsy is not routinely required but should be performed when lesions exhibit concerning features such as tenderness, bleeding, ulceration, rapid growth, marked hyperkeratosis, or failure to respond to standard treatment. Histopathology typically reveals atypical keratinocytes in the basal layers of the epidermis, with disordered maturation and parakeratosis.
Treatment depends on the number, size, location, and thickness of lesions. Isolated lesions are commonly treated with cryotherapy using liquid nitrogen. For patients with multiple lesions or field cancerization, topical therapies such as 5-fluorouracil, imiquimod, diclofenac gel, tirbanibulin ointment, or photodynamic therapy may be used. Procedural options include curettage, excision, chemical peels, dermabrasion, and laser therapy. Combination and sequential treatment approaches are frequently employed, particularly in refractory or extensive disease.
Referral to a dermatologist is recommended for patients with suspicious or recurrent lesions, extensive disease, immunosuppression, or when biopsy is indicated. Long-term follow-up is essential, typically on an annual or semiannual basis, due to the risk of progression to squamous cell carcinoma. Although the risk of transformation of an individual AK is low, the cumulative risk increases in patients with multiple lesions.
Prevention is a critical component of management. Measures include avoidance of excessive sun exposure, regular use of broad-spectrum sunscreens, protective clothing, and avoidance of tanning beds. Routine self-examination and regular professional skin evaluations are advised, especially for high-risk individuals. Preventive strategies such as nicotinamide supplementation and consistent sunscreen use have been shown to reduce the development of new actinic keratoses.
Actinic keratoses (AKs) are common premalignant skin lesions that typically present as multiple, ill-defined pink to yellow-brown, dry, scaly patches or plaques. They occur predominantly on chronically sun-exposed areas and are most often seen in middle-aged and elderly individuals. AKs are also known as solar keratoses or senile keratoses and are classified under ICD-10-CM code L57.0.
AKs are highly prevalent worldwide, with frequency varying by geographic location and sun exposure. In the northern hemisphere, approximately 11% to 25% of adults have at least one AK, whereas in regions closer to the equator, prevalence rises to 40% to 60%. In the United States, tens of millions of cases are estimated annually. The condition is more common in fair-skinned individuals, particularly those who burn rather than tan, and prevalence increases significantly with age. Men are affected more often than women, especially between the ages of 65 and 74 years.
Clinically, AKs appear on sun-damaged skin, most commonly involving the face, neck, scalp, ears, dorsal hands, and forearms. Early lesions may present as subtle erythematous patches with minimal scaling, while more advanced lesions develop thicker, yellowish, hyperkeratotic scales and feel rough or gritty on palpation. Lesions are usually less than 1 cm in diameter. Surrounding skin often shows signs of chronic photodamage, including atrophy, pigmentary changes, and telangiectasia. Several clinical variants exist, such as hypertrophic AKs with cutaneous horns, lichenoid AKs, proliferative AKs, pigmented AKs, and actinic cheilitis involving the lips.
The primary etiologic factor in the development of AKs is chronic ultraviolet (UV) radiation exposure. Additional contributing factors include ionizing radiation, arsenic exposure, polycyclic hydrocarbons, immunosuppression, human papillomavirus infection, cigarette smoke, and chronic skin injury. Genetic susceptibility also plays a role, particularly in conditions such as xeroderma pigmentosum and other inherited disorders associated with impaired DNA repair or increased skin cancer risk. Mutations in the p53 tumor suppressor gene are commonly implicated.
Diagnosis of actinic keratosis is usually clinical, based on characteristic appearance and patient history of sun exposure. A biopsy is not routinely required but should be performed when lesions exhibit concerning features such as tenderness, bleeding, ulceration, rapid growth, marked hyperkeratosis, or failure to respond to standard treatment. Histopathology typically reveals atypical keratinocytes in the basal layers of the epidermis, with disordered maturation and parakeratosis.
Treatment depends on the number, size, location, and thickness of lesions. Isolated lesions are commonly treated with cryotherapy using liquid nitrogen. For patients with multiple lesions or field cancerization, topical therapies such as 5-fluorouracil, imiquimod, diclofenac gel, tirbanibulin ointment, or photodynamic therapy may be used. Procedural options include curettage, excision, chemical peels, dermabrasion, and laser therapy. Combination and sequential treatment approaches are frequently employed, particularly in refractory or extensive disease.
Referral to a dermatologist is recommended for patients with suspicious or recurrent lesions, extensive disease, immunosuppression, or when biopsy is indicated. Long-term follow-up is essential, typically on an annual or semiannual basis, due to the risk of progression to squamous cell carcinoma. Although the risk of transformation of an individual AK is low, the cumulative risk increases in patients with multiple lesions.
Prevention is a critical component of management. Measures include avoidance of excessive sun exposure, regular use of broad-spectrum sunscreens, protective clothing, and avoidance of tanning beds. Routine self-examination and regular professional skin evaluations are advised, especially for high-risk individuals. Preventive strategies such as nicotinamide supplementation and consistent sunscreen use have been shown to reduce the development of new actinic keratoses.


- Published on
KembaraXtra-Medicine- Acute Bronchitis
Acute bronchitis is a self-limited inflammatory condition of the trachea and bronchi, most commonly referred to as a “chest cold.” It is characterized by transient inflammation of the lower airways and typically follows an upper respiratory tract infection. The condition usually resolves on its own without long-term sequelae.
Acute bronchitis is common, particularly during the winter months, and occurs most frequently in smokers, older adults, and young children. In the United States, cough accounts for nearly 30 million ambulatory visits each year, with more than 12 million resulting in a diagnosis of bronchitis. Acute lower respiratory tract infection remains the most common condition treated in primary care settings.
Clinically, acute bronchitis often begins with symptoms typical of a common cold, such as nasal congestion and sore throat, followed by the development of cough. The cough is usually worse in the morning and may be productive, largely due to transient bronchial hyperresponsiveness. Patients may also experience low-grade fever, substernal discomfort that worsens with coughing, postnasal drip, and pharyngeal erythema. On auscultation, rhonchi that clear with coughing and occasional wheezing may be present. Illness severity varies depending on host factors such as age, immune status, smoking history, and underlying medical conditions. While mild cases resolve within 7 to 10 days, cough may persist for up to 3 weeks or longer in some individuals.
The most common cause of acute bronchitis is viral infection. Frequently implicated viruses include rhinovirus, influenza virus, adenovirus, respiratory syncytial virus, parainfluenza virus, coronavirus, and human metapneumovirus. Less commonly, atypical organisms such as Mycoplasma pneumoniae and Chlamydia pneumoniae may be responsible. Bacterial causes are uncommon but include Bordetella pertussis, Haemophilus influenzae, Moraxella catarrhalis, and Streptococcus pneumoniae.
The diagnosis of acute bronchitis is primarily clinical. Important conditions to consider in the differential diagnosis include pneumonia, asthma, sinusitis, bronchiolitis, influenza, gastroesophageal reflux disease, congestive heart failure (particularly in older adults), medication-induced cough, and bronchogenic neoplasm. Diagnostic testing is seldom necessary unless there is concern for pneumonia, influenza, or another serious underlying condition.
Routine laboratory testing is generally not required. Chest radiography is reserved for patients with suspected pneumonia, those with chronic obstructive pulmonary disease who fail to improve, or individuals with concerning features such as high fever, hypoxia, or focal lung findings.
Management of acute bronchitis is largely supportive. Nonpharmacologic measures include avoidance of tobacco smoke and other pulmonary irritants, increased fluid intake, and the use of vaporizers or humidifiers to improve airway comfort. Treatment focuses on symptom relief rather than eradication of infection.
Pharmacologic therapy is aimed at controlling cough and wheezing. Inhaled bronchodilators such as albuterol may be used for 1 to 2 weeks in patients with wheezing or significant cough and have been shown to reduce cough duration in adults with uncomplicated disease. Cough suppressants such as dextromethorphan and expectorants like guaifenesin are commonly recommended. Codeine may be considered for severe cough that significantly disrupts sleep.
Antibiotics are generally not indicated for acute bronchitis and should be reserved for select cases, such as patients with suspected pertussis, confirmed atypical bacterial infection, or those with COPD exacerbations accompanied by increased dyspnea and purulent sputum. Despite evidence showing minimal benefit, antibiotics are prescribed in 70% to 90% of visits for acute bronchitis, contributing to antibiotic resistance and increased adverse effects, particularly gastrointestinal symptoms. Clinical trials have consistently demonstrated no meaningful difference in outcomes between antibiotic and placebo therapy in uncomplicated cases.
The prognosis for acute bronchitis is excellent. Most patients recover fully within 7 to 10 days, though cough commonly persists for 10 to 14 days after presentation. Patients should be reassured about the expected course of illness and advised to seek further evaluation if symptoms worsen or fail to improve.
Referral for pulmonary function testing is generally unnecessary and should be considered only in patients with recurrent episodes of bronchitis or suspected underlying chronic pulmonary disease. Patient education is essential, emphasizing that acute bronchitis is usually viral, antibiotics are unlikely to help, and symptomatic management is the cornerstone of care.
Acute bronchitis is a self-limited inflammatory condition of the trachea and bronchi, most commonly referred to as a “chest cold.” It is characterized by transient inflammation of the lower airways and typically follows an upper respiratory tract infection. The condition usually resolves on its own without long-term sequelae.
Acute bronchitis is common, particularly during the winter months, and occurs most frequently in smokers, older adults, and young children. In the United States, cough accounts for nearly 30 million ambulatory visits each year, with more than 12 million resulting in a diagnosis of bronchitis. Acute lower respiratory tract infection remains the most common condition treated in primary care settings.
Clinically, acute bronchitis often begins with symptoms typical of a common cold, such as nasal congestion and sore throat, followed by the development of cough. The cough is usually worse in the morning and may be productive, largely due to transient bronchial hyperresponsiveness. Patients may also experience low-grade fever, substernal discomfort that worsens with coughing, postnasal drip, and pharyngeal erythema. On auscultation, rhonchi that clear with coughing and occasional wheezing may be present. Illness severity varies depending on host factors such as age, immune status, smoking history, and underlying medical conditions. While mild cases resolve within 7 to 10 days, cough may persist for up to 3 weeks or longer in some individuals.
The most common cause of acute bronchitis is viral infection. Frequently implicated viruses include rhinovirus, influenza virus, adenovirus, respiratory syncytial virus, parainfluenza virus, coronavirus, and human metapneumovirus. Less commonly, atypical organisms such as Mycoplasma pneumoniae and Chlamydia pneumoniae may be responsible. Bacterial causes are uncommon but include Bordetella pertussis, Haemophilus influenzae, Moraxella catarrhalis, and Streptococcus pneumoniae.
The diagnosis of acute bronchitis is primarily clinical. Important conditions to consider in the differential diagnosis include pneumonia, asthma, sinusitis, bronchiolitis, influenza, gastroesophageal reflux disease, congestive heart failure (particularly in older adults), medication-induced cough, and bronchogenic neoplasm. Diagnostic testing is seldom necessary unless there is concern for pneumonia, influenza, or another serious underlying condition.
Routine laboratory testing is generally not required. Chest radiography is reserved for patients with suspected pneumonia, those with chronic obstructive pulmonary disease who fail to improve, or individuals with concerning features such as high fever, hypoxia, or focal lung findings.
Management of acute bronchitis is largely supportive. Nonpharmacologic measures include avoidance of tobacco smoke and other pulmonary irritants, increased fluid intake, and the use of vaporizers or humidifiers to improve airway comfort. Treatment focuses on symptom relief rather than eradication of infection.
Pharmacologic therapy is aimed at controlling cough and wheezing. Inhaled bronchodilators such as albuterol may be used for 1 to 2 weeks in patients with wheezing or significant cough and have been shown to reduce cough duration in adults with uncomplicated disease. Cough suppressants such as dextromethorphan and expectorants like guaifenesin are commonly recommended. Codeine may be considered for severe cough that significantly disrupts sleep.
Antibiotics are generally not indicated for acute bronchitis and should be reserved for select cases, such as patients with suspected pertussis, confirmed atypical bacterial infection, or those with COPD exacerbations accompanied by increased dyspnea and purulent sputum. Despite evidence showing minimal benefit, antibiotics are prescribed in 70% to 90% of visits for acute bronchitis, contributing to antibiotic resistance and increased adverse effects, particularly gastrointestinal symptoms. Clinical trials have consistently demonstrated no meaningful difference in outcomes between antibiotic and placebo therapy in uncomplicated cases.
The prognosis for acute bronchitis is excellent. Most patients recover fully within 7 to 10 days, though cough commonly persists for 10 to 14 days after presentation. Patients should be reassured about the expected course of illness and advised to seek further evaluation if symptoms worsen or fail to improve.
Referral for pulmonary function testing is generally unnecessary and should be considered only in patients with recurrent episodes of bronchitis or suspected underlying chronic pulmonary disease. Patient education is essential, emphasizing that acute bronchitis is usually viral, antibiotics are unlikely to help, and symptomatic management is the cornerstone of care.

- Published on
Emergency And Acute Medicine: Hyperthyroidism
Basics
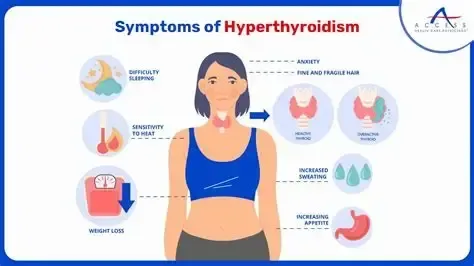
Description Hyperthyroidism is caused by excessive thyroid hormone production, producing a continuum of disease due to direct metabolic effects of thyroid hormones and increased sensitivity to catecholamines. Clinical states range from subclinical or mild hyperthyroidism to thyrotoxicosis and, in 1–2% of cases, life-threatening thyroid storm. Thyroid regulation occurs through hypothalamic TRH stimulating pituitary TSH release, which increases thyroidal secretion of T4 and T3. Most circulating hormone is T4, which is peripherally converted to the more biologically active T3. Genetic predisposition plays a role, particularly in Graves disease, which is associated with HLA-B8 and HLA-DR3, and in some familial cases of nontoxic goiter with autosomal dominant inheritance.
Etiology Primary hyperthyroidism includes Graves disease, toxic multinodular or uninodular goiter, iodine-induced hyperthyroidism, and thyroiditis (postpartum, radiation, subacute de Quervain, and chronic lymphocytic). Other causes include metastatic thyroid cancer, ectopic thyroid tissue such as struma ovarii, pituitary adenoma, drug-induced hyperthyroidism (amiodarone, lithium, interferon, interleukin-2, iodinated contrast), factitious thyrotoxicosis from excess hormone ingestion, and aspirin overdose.
Diagnosis
Alert Thyroid storm is a medical emergency and may be precipitated by infection, trauma, surgery, diabetic ketoacidosis, myocardial infarction, stroke, chemotherapy, organophosphate intoxication, or abrupt withdrawal of antithyroid medications.
Signs and symptoms Findings reflect heightened end-organ responsiveness to thyroid hormone. Common signs include fever, tachycardia with widened pulse pressure, diaphoresis, tremor, hyperreflexia, goiter, thyromegaly, thyroid bruit, exophthalmos, lid lag, pretibial myxedema, congestive heart failure, shock, and psychosis. Symptoms include weight loss despite increased appetite, palpitations, chest pain, heat intolerance, diarrhea, vomiting, weakness, anxiety, insomnia, menstrual irregularities, and fatigue. Thyroid storm presents with exaggerated manifestations including extreme tachyarrhythmias, heart failure, shock, delirium, coma, seizures, and thromboembolic events.
Geriatric considerations Apathetic hyperthyroidism often presents with subtle findings such as refractory atrial fibrillation, heart failure, weight loss, depression, tremor, and emotional lability rather than classic hyperadrenergic features.
History Typically reveals gradual onset of symptoms.
Physical exam May show fever, tachycardia, systolic hypertension with widened pulse pressure, tachypnea, alopecia, fine diaphoretic skin, irregularly irregular rhythm, lung rales, RUQ tenderness, jaundice, proximal muscle weakness, tremor, and altered mental status.
Essential workup Identify underlying cause and precipitating factors. Plasma TSH is the preferred initial test; a normal TSH generally excludes hyperthyroidism. Low TSH with normal T4 requires T3 measurement to rule out T3 thyrotoxicosis. If laboratory testing is delayed or unavailable, strong clinical suspicion should prompt treatment.
Diagnosis tests and interpretation
Laboratory TSH is typically suppressed, with elevated free T4. Approximately 5% of patients have isolated T3 thyrotoxicosis. Additional testing may include CBC, chemistry panel, liver enzymes, ABG, glucose, and cardiac markers to evaluate complications or precipitants.
Imaging Chest radiograph is useful in heart failure or infection.
Diagnostic procedures ECG commonly shows sinus tachycardia or new-onset atrial fibrillation and helps identify ischemia as a precipitating factor.
Differential diagnosis Pheochromocytoma, sepsis, sympathomimetic intoxication, psychosis, heat stroke, delirium tremens, malignant hyperthermia, neuroleptic malignant syndrome, serotonin syndrome, hypothalamic stroke, hypothyroidism with apathetic presentation, and factitious thyrotoxicosis.
Treatment
Prehospital Supportive care and stabilization.
Initial stabilization/therapy Manage airway, breathing, and circulation. Initiate cardiac monitoring, provide oxygen and IV fluids, and begin cooling measures. Use acetaminophen for fever and avoid aspirin, which increases free thyroid hormone levels.
Emergency department management Identify and treat precipitating causes. In suspected thyroid storm, initiate treatment based on clinical suspicion without delay. Inhibit hormone synthesis with thioamides, preferably propylthiouracil, which also reduces peripheral T4-to-T3 conversion; methimazole is an alternative. Block hormone release with iodine preparations only after thioamide administration and at least one hour later. Block peripheral effects using beta-blockers, with propranolol preferred due to inhibition of T4-to-T3 conversion; esmolol may be used when beta-1 selectivity is needed. Reduce peripheral conversion with corticosteroids. Adjunctive therapies include cholestyramine to reduce enterohepatic circulation of thyroid hormone and lithium if iodine is contraindicated. Manage heart failure, dehydration, hyperthermia, and associated conditions concurrently.
Medication Propylthiouracil, propranolol, iodine solutions (Lugol), methimazole, esmolol, hydrocortisone or dexamethasone, cholestyramine, lithium, guanethidine, and reserpine as indicated based on clinical context.
Pregnancy considerations Physiologic changes may mimic hyperthyroidism. Poorly controlled disease increases risks of hyperemesis gravidarum, preeclampsia, preterm labor, low birth weight, miscarriage, and stillbirth. PTU is preferred at the lowest effective dose; propranolol may be used cautiously. Radioactive iodine is contraindicated. Thyroidectomy is an option when medications are not tolerated. Postpartum thyroiditis occurs in up to 10% of patients and often resolves within one year.
Follow-up and disposition
Admission criteria Thyroid storm, need for IV rate control, or significant instability.
Discharge criteria Mild symptoms responsive to oral therapy with reliable follow-up.
Follow-up recommendations Arrange timely primary care or endocrinology follow-up.
Key points Thyroid storm is highly lethal without prompt treatment and often requires empiric therapy. Never administer iodine before blocking hormone synthesis with a thioamide. Radioactive iodine is contraindicated in pregnancy.
Basics
Description Hyperthyroidism is caused by excessive thyroid hormone production, producing a continuum of disease due to direct metabolic effects of thyroid hormones and increased sensitivity to catecholamines. Clinical states range from subclinical or mild hyperthyroidism to thyrotoxicosis and, in 1–2% of cases, life-threatening thyroid storm. Thyroid regulation occurs through hypothalamic TRH stimulating pituitary TSH release, which increases thyroidal secretion of T4 and T3. Most circulating hormone is T4, which is peripherally converted to the more biologically active T3. Genetic predisposition plays a role, particularly in Graves disease, which is associated with HLA-B8 and HLA-DR3, and in some familial cases of nontoxic goiter with autosomal dominant inheritance.
Etiology Primary hyperthyroidism includes Graves disease, toxic multinodular or uninodular goiter, iodine-induced hyperthyroidism, and thyroiditis (postpartum, radiation, subacute de Quervain, and chronic lymphocytic). Other causes include metastatic thyroid cancer, ectopic thyroid tissue such as struma ovarii, pituitary adenoma, drug-induced hyperthyroidism (amiodarone, lithium, interferon, interleukin-2, iodinated contrast), factitious thyrotoxicosis from excess hormone ingestion, and aspirin overdose.
Diagnosis
Alert Thyroid storm is a medical emergency and may be precipitated by infection, trauma, surgery, diabetic ketoacidosis, myocardial infarction, stroke, chemotherapy, organophosphate intoxication, or abrupt withdrawal of antithyroid medications.
Signs and symptoms Findings reflect heightened end-organ responsiveness to thyroid hormone. Common signs include fever, tachycardia with widened pulse pressure, diaphoresis, tremor, hyperreflexia, goiter, thyromegaly, thyroid bruit, exophthalmos, lid lag, pretibial myxedema, congestive heart failure, shock, and psychosis. Symptoms include weight loss despite increased appetite, palpitations, chest pain, heat intolerance, diarrhea, vomiting, weakness, anxiety, insomnia, menstrual irregularities, and fatigue. Thyroid storm presents with exaggerated manifestations including extreme tachyarrhythmias, heart failure, shock, delirium, coma, seizures, and thromboembolic events.
Geriatric considerations Apathetic hyperthyroidism often presents with subtle findings such as refractory atrial fibrillation, heart failure, weight loss, depression, tremor, and emotional lability rather than classic hyperadrenergic features.
History Typically reveals gradual onset of symptoms.
Physical exam May show fever, tachycardia, systolic hypertension with widened pulse pressure, tachypnea, alopecia, fine diaphoretic skin, irregularly irregular rhythm, lung rales, RUQ tenderness, jaundice, proximal muscle weakness, tremor, and altered mental status.
Essential workup Identify underlying cause and precipitating factors. Plasma TSH is the preferred initial test; a normal TSH generally excludes hyperthyroidism. Low TSH with normal T4 requires T3 measurement to rule out T3 thyrotoxicosis. If laboratory testing is delayed or unavailable, strong clinical suspicion should prompt treatment.
Diagnosis tests and interpretation
Laboratory TSH is typically suppressed, with elevated free T4. Approximately 5% of patients have isolated T3 thyrotoxicosis. Additional testing may include CBC, chemistry panel, liver enzymes, ABG, glucose, and cardiac markers to evaluate complications or precipitants.
Imaging Chest radiograph is useful in heart failure or infection.
Diagnostic procedures ECG commonly shows sinus tachycardia or new-onset atrial fibrillation and helps identify ischemia as a precipitating factor.
Differential diagnosis Pheochromocytoma, sepsis, sympathomimetic intoxication, psychosis, heat stroke, delirium tremens, malignant hyperthermia, neuroleptic malignant syndrome, serotonin syndrome, hypothalamic stroke, hypothyroidism with apathetic presentation, and factitious thyrotoxicosis.
Treatment
Prehospital Supportive care and stabilization.
Initial stabilization/therapy Manage airway, breathing, and circulation. Initiate cardiac monitoring, provide oxygen and IV fluids, and begin cooling measures. Use acetaminophen for fever and avoid aspirin, which increases free thyroid hormone levels.
Emergency department management Identify and treat precipitating causes. In suspected thyroid storm, initiate treatment based on clinical suspicion without delay. Inhibit hormone synthesis with thioamides, preferably propylthiouracil, which also reduces peripheral T4-to-T3 conversion; methimazole is an alternative. Block hormone release with iodine preparations only after thioamide administration and at least one hour later. Block peripheral effects using beta-blockers, with propranolol preferred due to inhibition of T4-to-T3 conversion; esmolol may be used when beta-1 selectivity is needed. Reduce peripheral conversion with corticosteroids. Adjunctive therapies include cholestyramine to reduce enterohepatic circulation of thyroid hormone and lithium if iodine is contraindicated. Manage heart failure, dehydration, hyperthermia, and associated conditions concurrently.
Medication Propylthiouracil, propranolol, iodine solutions (Lugol), methimazole, esmolol, hydrocortisone or dexamethasone, cholestyramine, lithium, guanethidine, and reserpine as indicated based on clinical context.
Pregnancy considerations Physiologic changes may mimic hyperthyroidism. Poorly controlled disease increases risks of hyperemesis gravidarum, preeclampsia, preterm labor, low birth weight, miscarriage, and stillbirth. PTU is preferred at the lowest effective dose; propranolol may be used cautiously. Radioactive iodine is contraindicated. Thyroidectomy is an option when medications are not tolerated. Postpartum thyroiditis occurs in up to 10% of patients and often resolves within one year.
Follow-up and disposition
Admission criteria Thyroid storm, need for IV rate control, or significant instability.
Discharge criteria Mild symptoms responsive to oral therapy with reliable follow-up.
Follow-up recommendations Arrange timely primary care or endocrinology follow-up.
Key points Thyroid storm is highly lethal without prompt treatment and often requires empiric therapy. Never administer iodine before blocking hormone synthesis with a thioamide. Radioactive iodine is contraindicated in pregnancy.
- Published on
Emergency And Acute Medicine: Hyperparathyroidism
Basics
Description Hyperparathyroidism is characterized by excess parathyroid hormone (PTH) leading to metabolic effects including decreased urinary calcium excretion, increased urinary phosphate loss, increased renal conversion of 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D, and increased calcium and phosphate release from bone. Hypercalcemia is the primary metabolic abnormality. Despite reduced renal calcium excretion, hypercalciuria occurs due to elevated serum calcium, often accompanied by urinary magnesium loss. Magnesium is essential for both PTH secretion and peripheral PTH action; hypomagnesemia may blunt PTH effects. Genetic associations include multiple endocrine neoplasia (MEN) syndromes: MEN 1 (hyperparathyroidism, pancreatic islet tumors, pituitary disease) and MEN 2 (hyperparathyroidism in MEN 2A, medullary thyroid carcinoma, pheochromocytoma, and mucosal neuromas in MEN 2B).
Etiology Primary hyperparathyroidism results from parathyroid adenoma (≈85%), hyperplasia (≈14%), or rarely carcinoma (<1%). Secondary hyperparathyroidism is a compensatory response to vitamin D deficiency or chronic kidney disease with hyperphosphatemia; calcium levels are low or normal with elevated PTH.
Diagnosis
Signs and symptoms Classic features include renal stones, bone pain, gastrointestinal complaints, and neuropsychiatric changes. Alert Hypercalcemic crisis presents with anorexia, nausea, vomiting, and progressive mental status depression.
History Symptoms correlate with severity and rapidity of hypercalcemia.
Pediatric considerations Neonates born to hypoparathyroid mothers may present with hypotonia, weakness, and lethargy; hypercalcemic infants may have distinctive facial features including broad forehead, epicanthal folds, underdeveloped nasal bridge, and prominent upper lip.
Physical exam Findings may include dehydration, hypertension despite volume depletion, cardiac conduction abnormalities (bradyarrhythmias, bundle branch block, complete heart block, asystole), shortened QT interval, potentiation of digoxin toxicity, neurologic deficits (decreased reflexes, proximal weakness, lethargy, coma), psychiatric symptoms (depression, anxiety, psychosis), gastrointestinal symptoms (anorexia, constipation, peptic ulcer disease, pancreatitis), musculoskeletal pain, gout or pseudogout, and renal manifestations including nephrolithiasis and nephrocalcinosis.
Essential workup Measure serum calcium and albumin to assess corrected calcium. Evaluate for symptoms of severe hypercalcemia or impending parathyroid crisis. If asymptomatic with normal ECG and corrected calcium <14 mg/dL, no further ED testing is required. If symptomatic or calcium ≥14 mg/dL, obtain ionized calcium, electrolytes, BUN/creatinine, phosphorus, magnesium, alkaline phosphatase, ESR, TSH, CBC, and chest radiograph.
Diagnosis tests and interpretation
Laboratory Correct calcium for albumin: corrected Ca (mg/dL) = measured Ca + 0.8 × (4 − albumin). Acidosis increases ionized calcium by reducing albumin binding. Phosphorus is typically low in primary hyperparathyroidism and elevated in secondary disease. Chloride-to-phosphate ratio >33 favors hyperparathyroidism; <30 suggests malignancy. Alkaline phosphatase is elevated in ~50% of cases. ESR and anemia are usually normal in hyperparathyroidism but elevated in malignancy or granulomatous disease. Magnesium is often low or low-normal. PTH is elevated in primary and secondary hyperparathyroidism. PTH-related peptide suggests malignancy-associated hypercalcemia.
Imaging Chest radiograph evaluates volume status and screens for malignancy or granulomatous disease.
Diagnostic procedures Definitive diagnosis and treatment are established with parathyroidectomy.
Differential diagnosis PTH-mediated causes include primary or secondary hyperparathyroidism and familial hypocalciuric hypercalcemia. Non-PTH causes include malignancy, vitamin D excess or granulomatous disease, immobilization (e.g., Paget disease), and drug-induced hypercalcemia (thiazides, lithium, vitamin A, aluminum antacids, estrogens, androgens, tamoxifen).
Treatment
Prehospital May present primarily with psychiatric manifestations.
Initial stabilization/therapy Place on cardiac monitor if symptomatic or calcium >14 mg/dL. Begin IV hydration with 0.9% normal saline and correct acidosis.
Emergency department management Treat hypercalcemia with aggressive isotonic saline hydration (≥250 mL/hr unless limited by heart failure) targeting urine output ≥100 mL/hr. After adequate hydration, add loop diuretics to enhance calciuresis; avoid thiazides. Consider glucocorticoids in vitamin D–mediated or granulomatous disease. Initiate bisphosphonates in coordination with endocrinology. Treat dysrhythmias conventionally and use extreme caution with digoxin. Stop contributing medications and monitor closely for heart failure and electrolyte disturbances. Use calcitonin when hydration is contraindicated and initiate dialysis for refractory hypercalcemia with renal failure.
Medication First line Normal saline infusion; furosemide 40 mg IV q2–4h after rehydration; prednisone 40–60 mg PO or hydrocortisone 100 mg IV. Second line (with endocrinology) Calcitonin salmon 4 U/kg SC q12h; pamidronate 60–90 mg IV depending on calcium level; zoledronic acid 4 mg IV; cinacalcet 30 mg PO daily or BID for secondary hyperparathyroidism or carcinoma.
Follow-up and disposition
Admission criteria Corrected calcium >14 mg/dL, symptomatic hypercalcemia, or cardiac conduction abnormalities.
Discharge criteria Asymptomatic patients with calcium <14 mg/dL who can maintain hydration.
Issues for referral Endocrinology referral for PTH evaluation and definitive management.
Follow-up recommendations Arrange outpatient PTH testing, reinforce hydration, and discontinue medications that increase calcium levels.
Key points Hyperparathyroid-related hypercalcemia is often mild and rarely exceeds 14 mg/dL; higher levels suggest malignancy. Measure ionized or albumin-corrected calcium. Administering loop diuretics before adequate hydration worsens hypercalcemia and is a common management error.
Basics
Description Hyperparathyroidism is characterized by excess parathyroid hormone (PTH) leading to metabolic effects including decreased urinary calcium excretion, increased urinary phosphate loss, increased renal conversion of 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D, and increased calcium and phosphate release from bone. Hypercalcemia is the primary metabolic abnormality. Despite reduced renal calcium excretion, hypercalciuria occurs due to elevated serum calcium, often accompanied by urinary magnesium loss. Magnesium is essential for both PTH secretion and peripheral PTH action; hypomagnesemia may blunt PTH effects. Genetic associations include multiple endocrine neoplasia (MEN) syndromes: MEN 1 (hyperparathyroidism, pancreatic islet tumors, pituitary disease) and MEN 2 (hyperparathyroidism in MEN 2A, medullary thyroid carcinoma, pheochromocytoma, and mucosal neuromas in MEN 2B).
Etiology Primary hyperparathyroidism results from parathyroid adenoma (≈85%), hyperplasia (≈14%), or rarely carcinoma (<1%). Secondary hyperparathyroidism is a compensatory response to vitamin D deficiency or chronic kidney disease with hyperphosphatemia; calcium levels are low or normal with elevated PTH.
Diagnosis
Signs and symptoms Classic features include renal stones, bone pain, gastrointestinal complaints, and neuropsychiatric changes. Alert Hypercalcemic crisis presents with anorexia, nausea, vomiting, and progressive mental status depression.
History Symptoms correlate with severity and rapidity of hypercalcemia.
Pediatric considerations Neonates born to hypoparathyroid mothers may present with hypotonia, weakness, and lethargy; hypercalcemic infants may have distinctive facial features including broad forehead, epicanthal folds, underdeveloped nasal bridge, and prominent upper lip.
Physical exam Findings may include dehydration, hypertension despite volume depletion, cardiac conduction abnormalities (bradyarrhythmias, bundle branch block, complete heart block, asystole), shortened QT interval, potentiation of digoxin toxicity, neurologic deficits (decreased reflexes, proximal weakness, lethargy, coma), psychiatric symptoms (depression, anxiety, psychosis), gastrointestinal symptoms (anorexia, constipation, peptic ulcer disease, pancreatitis), musculoskeletal pain, gout or pseudogout, and renal manifestations including nephrolithiasis and nephrocalcinosis.
Essential workup Measure serum calcium and albumin to assess corrected calcium. Evaluate for symptoms of severe hypercalcemia or impending parathyroid crisis. If asymptomatic with normal ECG and corrected calcium <14 mg/dL, no further ED testing is required. If symptomatic or calcium ≥14 mg/dL, obtain ionized calcium, electrolytes, BUN/creatinine, phosphorus, magnesium, alkaline phosphatase, ESR, TSH, CBC, and chest radiograph.
Diagnosis tests and interpretation
Laboratory Correct calcium for albumin: corrected Ca (mg/dL) = measured Ca + 0.8 × (4 − albumin). Acidosis increases ionized calcium by reducing albumin binding. Phosphorus is typically low in primary hyperparathyroidism and elevated in secondary disease. Chloride-to-phosphate ratio >33 favors hyperparathyroidism; <30 suggests malignancy. Alkaline phosphatase is elevated in ~50% of cases. ESR and anemia are usually normal in hyperparathyroidism but elevated in malignancy or granulomatous disease. Magnesium is often low or low-normal. PTH is elevated in primary and secondary hyperparathyroidism. PTH-related peptide suggests malignancy-associated hypercalcemia.
Imaging Chest radiograph evaluates volume status and screens for malignancy or granulomatous disease.
Diagnostic procedures Definitive diagnosis and treatment are established with parathyroidectomy.
Differential diagnosis PTH-mediated causes include primary or secondary hyperparathyroidism and familial hypocalciuric hypercalcemia. Non-PTH causes include malignancy, vitamin D excess or granulomatous disease, immobilization (e.g., Paget disease), and drug-induced hypercalcemia (thiazides, lithium, vitamin A, aluminum antacids, estrogens, androgens, tamoxifen).
Treatment
Prehospital May present primarily with psychiatric manifestations.
Initial stabilization/therapy Place on cardiac monitor if symptomatic or calcium >14 mg/dL. Begin IV hydration with 0.9% normal saline and correct acidosis.
Emergency department management Treat hypercalcemia with aggressive isotonic saline hydration (≥250 mL/hr unless limited by heart failure) targeting urine output ≥100 mL/hr. After adequate hydration, add loop diuretics to enhance calciuresis; avoid thiazides. Consider glucocorticoids in vitamin D–mediated or granulomatous disease. Initiate bisphosphonates in coordination with endocrinology. Treat dysrhythmias conventionally and use extreme caution with digoxin. Stop contributing medications and monitor closely for heart failure and electrolyte disturbances. Use calcitonin when hydration is contraindicated and initiate dialysis for refractory hypercalcemia with renal failure.
Medication First line Normal saline infusion; furosemide 40 mg IV q2–4h after rehydration; prednisone 40–60 mg PO or hydrocortisone 100 mg IV. Second line (with endocrinology) Calcitonin salmon 4 U/kg SC q12h; pamidronate 60–90 mg IV depending on calcium level; zoledronic acid 4 mg IV; cinacalcet 30 mg PO daily or BID for secondary hyperparathyroidism or carcinoma.
Follow-up and disposition
Admission criteria Corrected calcium >14 mg/dL, symptomatic hypercalcemia, or cardiac conduction abnormalities.
Discharge criteria Asymptomatic patients with calcium <14 mg/dL who can maintain hydration.
Issues for referral Endocrinology referral for PTH evaluation and definitive management.
Follow-up recommendations Arrange outpatient PTH testing, reinforce hydration, and discontinue medications that increase calcium levels.
Key points Hyperparathyroid-related hypercalcemia is often mild and rarely exceeds 14 mg/dL; higher levels suggest malignancy. Measure ionized or albumin-corrected calcium. Administering loop diuretics before adequate hydration worsens hypercalcemia and is a common management error.
- Published on
Emergency And Acute Medicine: Hypertensive Emergencies
Basics
Description Hypertensive crisis refers to severe blood pressure elevation, typically SBP >179 mm Hg or DBP >109 mm Hg. Hypertensive urgency is severe BP elevation without acute end-organ injury. Hypertensive emergency is severe BP elevation with acute target-organ damage. In hypertensive emergency, autoregulation fails: arterioles initially constrict to protect capillary beds, but extreme pressures overwhelm this response, causing endothelial injury, increased permeability, platelet and coagulation activation, and fibrin deposition. Sympathetic and renin–angiotensin activation further increase vasoconstriction and inflammation, worsening ischemia and perpetuating a vicious cycle. Commonly affected organs include brain (encephalopathy, ischemic stroke, intracerebral hemorrhage), retina (hemorrhage, papilledema), heart (ACS, acute heart failure, aortic dissection), kidneys (acute renal failure), and placenta (preeclampsia/eclampsia).
Etiology Causes include essential hypertension; renal vascular or parenchymal disease; coarctation of the aorta; CNS injury (head trauma, stroke/ICH, tumor, spinal cord injury); endocrine disorders (pheochromocytoma, Cushing syndrome, primary hyperaldosteronism, renin-secreting tumors); drugs and toxins (cocaine, PCP, amphetamines, erythropoietin, tacrolimus, cyclosporine, corticosteroids, oral contraceptives, MAOI interactions, lead); withdrawal of antihypertensives; autonomic hyperreactivity (Guillain–Barré, acute intermittent porphyria); postoperative pain or anesthetic complications; and pregnancy-related disease (preeclampsia/eclampsia).
Diagnosis
History Review prescribed and OTC medications, antihypertensive adherence or withdrawal, duration and control of baseline hypertension, prior end-organ disease, comorbidities (CAD, diabetes, obesity), and recreational drug use. Screen for end-organ injury, commonly dyspnea, chest pain, headache, confusion, and focal neurologic deficits.
Physical exam Measure BP in both arms using appropriate cuff size. Assess for neurologic deficits and mental status changes, perform funduscopic exam for hemorrhages or papilledema, evaluate cardiovascular status (JVP, crackles, S3, aortic regurg murmur), and check for pulse asymmetry.
Essential workup Obtain a 12-lead ECG for ischemia and LVH, and assess kidney function because acute renal failure may be clinically silent.
Diagnosis tests and interpretation
Laboratory CBC (anemia and thrombocytopenia may indicate thrombotic microangiopathy), BMP including BUN/creatinine and electrolytes (hypokalemia suggests mineralocorticoid excess), urinalysis (protein, blood, casts), urine toxicology when drug use is suspected, and pregnancy test when appropriate. Use standard chest pain protocols when indicated.
Imaging Chest radiograph for cardiopulmonary symptoms, head CT for headache, confusion, or focal deficits, and CTA chest/abdomen if aortic dissection is suspected.
Procedures Consider arterial line monitoring in unstable cases. Lumbar puncture may be used to evaluate for subarachnoid hemorrhage after appropriate imaging.
Differential diagnosis ACS, acute heart failure, aortic dissection, ischemic stroke, intracerebral hemorrhage, subarachnoid hemorrhage, preeclampsia/eclampsia, withdrawal syndromes (beta-blocker, clonidine), and catecholamine excess states (pheochromocytoma, sympathomimetic intoxication, tyramine reaction with MAOIs).
Treatment
Prehospital Support ABCs and consider cautious BP reduction only when clearly indicated.
Initial stabilization/therapy Provide oxygen as needed, establish IV access, monitor ECG and pulse oximetry.
Emergency department management Hypertensive urgency does not require IV reduction; give missed home medications, use oral agents only, and lower BP gradually over 24–48 hours with close follow-up. Hypertensive emergency requires IV therapy guided by the end-organ injury rather than the absolute BP. Reduce MAP by no more than 20–25% in the first hour, then target roughly SBP ~160 and DBP ~100 over 2–6 hours, transitioning to oral agents within 6–12 hours once stable. Lower more gradually in acute CNS injury, but more rapidly in aortic dissection. Hypertensive encephalopathy: lower MAP up to 20% or DBP to 100–110 within the first hour, then normalize over 48–72 hours using nicardipine, clevidipine, or labetalol. Ischemic stroke: treat only if SBP >220 or DBP >120, or if thrombolysis planned (target <185/110 pre-tPA, then <180/105); avoid reducing MAP more than 15–20% in the first 24 hours; preferred agents are nicardipine, clevidipine, or labetalol. ICH/SAH: treat if SBP >180 or DBP >100; aim SBP 140–160 or MAP down 20–25% in the first hour; avoid nitroglycerin and nitroprusside due to cerebral vasodilation and ICP effects; use nicardipine, clevidipine, or labetalol. ACS: target MAP 60–100 using labetalol or esmolol with nitroglycerin; avoid hydralazine and nitroprusside. Acute heart failure: target MAP 60–100 with nitroprusside or nitroglycerin plus ACE inhibitor and/or loop diuretic. Acute renal failure or microangiopathy: reduce MAP 20–25% in the first hour using nicardipine, clevidipine, or fenoldopam; ACE inhibitors are preferred for scleroderma renal crisis. Aortic dissection: reduce shear force by lowering HR and BP; beta-blockade must come first; target SBP 100–120 and HR <65 within 20 minutes using esmolol plus a vasodilator (dihydropyridine CCB or nitroprusside); obtain urgent surgical consultation for type A.
Sympathomimetic crisis: avoid pure beta-blockade; use phentolamine or calcium channel blocker plus benzodiazepines; use clonidine for clonidine withdrawal.
Pregnancy considerations Preeclampsia is SBP >140 or DBP >90 with proteinuria after 20 weeks to 4 weeks postpartum, often with headache, visual changes, edema, or RUQ pain; target SBP 130–150 and DBP 80–100 using labetalol, nicardipine, hydralazine, and magnesium, with obstetric consultation.
Medication Clevidipine 1–16 mg/h infusion; nicardipine 2–15 mg/h infusion; labetalol 20–80 mg IV q10 min (max 300 mg) or 0.5–2 mg/min infusion; esmolol 80 mg IV bolus then 150 μg/kg/min infusion; nitroglycerin 5–100 μg/min infusion; nitroprusside 0.25–10 μg/kg/min infusion; hydralazine 10–20 mg IV bolus; fenoldopam 0.1–0.6 μg/kg/min infusion; enalaprilat 1.25–5 mg IV q6h; phentolamine 5–15 mg IV q5–15 min.
Follow-up and disposition
Admission criteria Any patient with acute end-organ injury requires admission; ICU monitoring is indicated for hypertensive emergency.
Discharge criteria No end-organ injury, reliable follow-up, known hypertension, reversible trigger such as medication nonadherence, and ability to resume a safe outpatient regimen; provide strict return precautions for chest pain, neurologic symptoms, or severe headache.
Follow-up recommendations Ensure timely primary care follow-up for medication initiation or adjustment and long-term BP control.
Key points Do not use IV agents for hypertensive urgency. In hypertensive emergency, the initial goal is controlled MAP reduction of 20–25% in the first hour, except for ischemic stroke and aortic dissection where targets differ. Avoid precipitous BP drops that can worsen ischemia. In aortic dissection prevent reflex tachycardia by starting beta-blockade first. In catecholamine excess avoid unopposed alpha stimulation by avoiding pure beta-blockers.
Basics
Description Hypertensive crisis refers to severe blood pressure elevation, typically SBP >179 mm Hg or DBP >109 mm Hg. Hypertensive urgency is severe BP elevation without acute end-organ injury. Hypertensive emergency is severe BP elevation with acute target-organ damage. In hypertensive emergency, autoregulation fails: arterioles initially constrict to protect capillary beds, but extreme pressures overwhelm this response, causing endothelial injury, increased permeability, platelet and coagulation activation, and fibrin deposition. Sympathetic and renin–angiotensin activation further increase vasoconstriction and inflammation, worsening ischemia and perpetuating a vicious cycle. Commonly affected organs include brain (encephalopathy, ischemic stroke, intracerebral hemorrhage), retina (hemorrhage, papilledema), heart (ACS, acute heart failure, aortic dissection), kidneys (acute renal failure), and placenta (preeclampsia/eclampsia).
Etiology Causes include essential hypertension; renal vascular or parenchymal disease; coarctation of the aorta; CNS injury (head trauma, stroke/ICH, tumor, spinal cord injury); endocrine disorders (pheochromocytoma, Cushing syndrome, primary hyperaldosteronism, renin-secreting tumors); drugs and toxins (cocaine, PCP, amphetamines, erythropoietin, tacrolimus, cyclosporine, corticosteroids, oral contraceptives, MAOI interactions, lead); withdrawal of antihypertensives; autonomic hyperreactivity (Guillain–Barré, acute intermittent porphyria); postoperative pain or anesthetic complications; and pregnancy-related disease (preeclampsia/eclampsia).
Diagnosis
History Review prescribed and OTC medications, antihypertensive adherence or withdrawal, duration and control of baseline hypertension, prior end-organ disease, comorbidities (CAD, diabetes, obesity), and recreational drug use. Screen for end-organ injury, commonly dyspnea, chest pain, headache, confusion, and focal neurologic deficits.
Physical exam Measure BP in both arms using appropriate cuff size. Assess for neurologic deficits and mental status changes, perform funduscopic exam for hemorrhages or papilledema, evaluate cardiovascular status (JVP, crackles, S3, aortic regurg murmur), and check for pulse asymmetry.
Essential workup Obtain a 12-lead ECG for ischemia and LVH, and assess kidney function because acute renal failure may be clinically silent.
Diagnosis tests and interpretation
Laboratory CBC (anemia and thrombocytopenia may indicate thrombotic microangiopathy), BMP including BUN/creatinine and electrolytes (hypokalemia suggests mineralocorticoid excess), urinalysis (protein, blood, casts), urine toxicology when drug use is suspected, and pregnancy test when appropriate. Use standard chest pain protocols when indicated.
Imaging Chest radiograph for cardiopulmonary symptoms, head CT for headache, confusion, or focal deficits, and CTA chest/abdomen if aortic dissection is suspected.
Procedures Consider arterial line monitoring in unstable cases. Lumbar puncture may be used to evaluate for subarachnoid hemorrhage after appropriate imaging.
Differential diagnosis ACS, acute heart failure, aortic dissection, ischemic stroke, intracerebral hemorrhage, subarachnoid hemorrhage, preeclampsia/eclampsia, withdrawal syndromes (beta-blocker, clonidine), and catecholamine excess states (pheochromocytoma, sympathomimetic intoxication, tyramine reaction with MAOIs).
Treatment
Prehospital Support ABCs and consider cautious BP reduction only when clearly indicated.
Initial stabilization/therapy Provide oxygen as needed, establish IV access, monitor ECG and pulse oximetry.
Emergency department management Hypertensive urgency does not require IV reduction; give missed home medications, use oral agents only, and lower BP gradually over 24–48 hours with close follow-up. Hypertensive emergency requires IV therapy guided by the end-organ injury rather than the absolute BP. Reduce MAP by no more than 20–25% in the first hour, then target roughly SBP ~160 and DBP ~100 over 2–6 hours, transitioning to oral agents within 6–12 hours once stable. Lower more gradually in acute CNS injury, but more rapidly in aortic dissection. Hypertensive encephalopathy: lower MAP up to 20% or DBP to 100–110 within the first hour, then normalize over 48–72 hours using nicardipine, clevidipine, or labetalol. Ischemic stroke: treat only if SBP >220 or DBP >120, or if thrombolysis planned (target <185/110 pre-tPA, then <180/105); avoid reducing MAP more than 15–20% in the first 24 hours; preferred agents are nicardipine, clevidipine, or labetalol. ICH/SAH: treat if SBP >180 or DBP >100; aim SBP 140–160 or MAP down 20–25% in the first hour; avoid nitroglycerin and nitroprusside due to cerebral vasodilation and ICP effects; use nicardipine, clevidipine, or labetalol. ACS: target MAP 60–100 using labetalol or esmolol with nitroglycerin; avoid hydralazine and nitroprusside. Acute heart failure: target MAP 60–100 with nitroprusside or nitroglycerin plus ACE inhibitor and/or loop diuretic. Acute renal failure or microangiopathy: reduce MAP 20–25% in the first hour using nicardipine, clevidipine, or fenoldopam; ACE inhibitors are preferred for scleroderma renal crisis. Aortic dissection: reduce shear force by lowering HR and BP; beta-blockade must come first; target SBP 100–120 and HR <65 within 20 minutes using esmolol plus a vasodilator (dihydropyridine CCB or nitroprusside); obtain urgent surgical consultation for type A.
Sympathomimetic crisis: avoid pure beta-blockade; use phentolamine or calcium channel blocker plus benzodiazepines; use clonidine for clonidine withdrawal.
Pregnancy considerations Preeclampsia is SBP >140 or DBP >90 with proteinuria after 20 weeks to 4 weeks postpartum, often with headache, visual changes, edema, or RUQ pain; target SBP 130–150 and DBP 80–100 using labetalol, nicardipine, hydralazine, and magnesium, with obstetric consultation.
Medication Clevidipine 1–16 mg/h infusion; nicardipine 2–15 mg/h infusion; labetalol 20–80 mg IV q10 min (max 300 mg) or 0.5–2 mg/min infusion; esmolol 80 mg IV bolus then 150 μg/kg/min infusion; nitroglycerin 5–100 μg/min infusion; nitroprusside 0.25–10 μg/kg/min infusion; hydralazine 10–20 mg IV bolus; fenoldopam 0.1–0.6 μg/kg/min infusion; enalaprilat 1.25–5 mg IV q6h; phentolamine 5–15 mg IV q5–15 min.
Follow-up and disposition
Admission criteria Any patient with acute end-organ injury requires admission; ICU monitoring is indicated for hypertensive emergency.
Discharge criteria No end-organ injury, reliable follow-up, known hypertension, reversible trigger such as medication nonadherence, and ability to resume a safe outpatient regimen; provide strict return precautions for chest pain, neurologic symptoms, or severe headache.
Follow-up recommendations Ensure timely primary care follow-up for medication initiation or adjustment and long-term BP control.
Key points Do not use IV agents for hypertensive urgency. In hypertensive emergency, the initial goal is controlled MAP reduction of 20–25% in the first hour, except for ischemic stroke and aortic dissection where targets differ. Avoid precipitous BP drops that can worsen ischemia. In aortic dissection prevent reflex tachycardia by starting beta-blockade first. In catecholamine excess avoid unopposed alpha stimulation by avoiding pure beta-blockers.
- Published on
Emergency And Acute Medicine: Hyperthermia
Basics
Description Hyperthermia represents a spectrum of heat-related illness caused by progressively overwhelming heat stress, ranging from dehydration and electrolyte abnormalities to thermoregulatory failure and multisystem organ dysfunction. Normal body temperature is maintained by balancing heat production and dissipation. At temperatures above 42°C (108°F), oxidative phosphorylation becomes uncoupled and critical enzymes fail. Heat stroke is defined by a core temperature >105°F (40.5°C) with failure of thermoregulation, severe CNS dysfunction, and multisystem organ failure. Classic heat stroke is nonexertional, typically affecting the elderly, very young, or debilitated patients who cannot escape a hot environment; it develops over days to weeks and is associated with severe dehydration and hot, often dry skin. Exertional heat stroke occurs in younger, healthy individuals under intense physical exertion, develops over hours, and may occur despite ongoing sweating. Heat exhaustion involves moderate temperature elevation, usually <104°F (40°C), preserved thermoregulation and CNS function, and fluid or salt depletion; without treatment it may progress to heat stroke.
Etiology Predisposing factors impair heat dissipation or increase heat production. Medical conditions include extremes of age, dehydration, cardiovascular disease, obesity, diabetes, hyperthyroidism, pheochromocytoma, febrile illness, and skin disorders limiting sweating. Pharmacologic contributors include sympathomimetics, cocaine, PCP, LSD, MAO inhibitors, antipsychotics, anxiolytics, anticholinergics, antihistamines, beta-blockers, diuretics, laxatives, and drug or alcohol withdrawal. Environmental risks include high heat and humidity, prolonged exertion, immobility, lack of air conditioning, poor acclimatization, and occlusive clothing.
Pediatric considerations Children are at increased risk due to higher body surface area–to–mass ratio and reduced sweating capacity.
Diagnosis
Signs and symptoms Heat stroke presents with the classic triad of hyperthermia, CNS dysfunction, and hot skin. Core temperature exceeds 105°F (40.5°C). Neurologic findings include delirium, coma, seizures, ataxia, and confusion. Cardiovascular findings include tachycardia, hypotension, wide pulse pressure, and conduction abnormalities. Pulmonary findings include tachypnea, respiratory alkalosis, hypoxemia, and noncardiogenic pulmonary edema. GI symptoms include nausea, vomiting, and diarrhea. Renal failure, rhabdomyolysis, hepatic failure with extreme transaminase elevation, and coagulopathy including DIC may occur and indicate poor prognosis. Heat exhaustion causes headache, fatigue, malaise, impaired judgment, nausea, vomiting, tachycardia, dehydration, tachypnea, and profuse sweating without severe CNS dysfunction. Heat cramps cause painful muscle cramps after heavy sweating and hypotonic fluid replacement, leading to hyponatremia. Heat edema presents as dependent lower-extremity swelling in nonacclimatized individuals. Heat syncope presents as transient loss of consciousness during heat exposure, especially in the elderly. Prickly heat causes a pruritic maculopapular or vesicular rash due to sweat duct obstruction.
Essential workup Measure accurate core temperature using a rectal or esophageal probe. Establish history of heat exposure. Heat exhaustion is a diagnosis of exclusion. Heat stroke requires both core temperature >40.5°C and severe CNS dysfunction.
Diagnosis tests and interpretation
Laboratory Obtain CBC (leukocytosis, hemoconcentration), electrolytes, BUN, creatinine, glucose, urinalysis (myoglobin), creatine kinase for rhabdomyolysis, ABG (acidosis and elevated lactate common in exertional heat stroke), blood and urine cultures if sepsis is considered, toxicology screen, PT/PTT and DIC panel, liver function tests, and troponin (elevation suggests poor prognosis).
Imaging ECG in elderly or cardiac risk patients. Chest radiograph for ARDS or aspiration. Head CT for altered mental status. Lumbar puncture may be required to exclude CNS infection.
Differential diagnosis Sepsis, thyroid storm, pheochromocytoma, stimulant or anticholinergic toxicity, meningitis or encephalitis, cerebral malaria, delirium tremens, neuroleptic malignant syndrome, malignant hyperthermia, and serotonin syndrome.
Treatment
Prehospital Remove patient from heat source, disrobe, and initiate cooling with wet sheets.
Initial stabilization/therapy Ensure airway, breathing, and circulation. Begin continuous core temperature monitoring. Initiate rapid cooling if temperature >104°F (40°C). Give IV 0.9% normal saline bolus if hypotensive. For altered mental status, administer glucose, thiamine, and naloxone as indicated.
Emergency department management Cooling is the priority. Use evaporative cooling with misted warm water and high airflow; combine with ice packs to groin and axilla. Cold-water immersion is effective but often impractical. Advanced techniques such as cold peritoneal lavage, extracorporeal circulation, or cold dialysis may be considered for refractory cases. Stop cooling at 102°F (39°C) to prevent hypothermia. Antipyretics are ineffective and alcohol sponge baths should be avoided. Supportive care includes aggressive isotonic fluid resuscitation while avoiding fluid overload. Insert Foley catheter to monitor urine output; target >2 mL/kg/hr if rhabdomyolysis is present. Treat seizures, agitation, and shivering with benzodiazepines. Avoid vasopressors and antiarrhythmics until adequate cooling, as many arrhythmias resolve with temperature normalization. Treat associated heat syndromes with appropriate hydration, electrolyte replacement, elevation or compression, and topical therapy as indicated.
Medication Benzodiazepines such as diazepam or lorazepam for seizures or agitation; naloxone when opioid toxicity is suspected.
Follow-up and disposition
Admission criteria All patients with heat stroke require ICU admission. Heat exhaustion requires admission if severe electrolyte abnormalities, renal failure, rhabdomyolysis, or advanced age are present.
Discharge criteria Patients without heat stroke or severe heat exhaustion who normalize clinically after treatment may be discharged with counseling.
Key points Heat stroke cannot be diagnosed without both core temperature >40.5°C and severe CNS dysfunction. Rapid cooling and supportive care are the cornerstones of management. Continuous core temperature monitoring is standard of care, and evaporative cooling is the preferred first-line method.
Basics
Description Hyperthermia represents a spectrum of heat-related illness caused by progressively overwhelming heat stress, ranging from dehydration and electrolyte abnormalities to thermoregulatory failure and multisystem organ dysfunction. Normal body temperature is maintained by balancing heat production and dissipation. At temperatures above 42°C (108°F), oxidative phosphorylation becomes uncoupled and critical enzymes fail. Heat stroke is defined by a core temperature >105°F (40.5°C) with failure of thermoregulation, severe CNS dysfunction, and multisystem organ failure. Classic heat stroke is nonexertional, typically affecting the elderly, very young, or debilitated patients who cannot escape a hot environment; it develops over days to weeks and is associated with severe dehydration and hot, often dry skin. Exertional heat stroke occurs in younger, healthy individuals under intense physical exertion, develops over hours, and may occur despite ongoing sweating. Heat exhaustion involves moderate temperature elevation, usually <104°F (40°C), preserved thermoregulation and CNS function, and fluid or salt depletion; without treatment it may progress to heat stroke.
Etiology Predisposing factors impair heat dissipation or increase heat production. Medical conditions include extremes of age, dehydration, cardiovascular disease, obesity, diabetes, hyperthyroidism, pheochromocytoma, febrile illness, and skin disorders limiting sweating. Pharmacologic contributors include sympathomimetics, cocaine, PCP, LSD, MAO inhibitors, antipsychotics, anxiolytics, anticholinergics, antihistamines, beta-blockers, diuretics, laxatives, and drug or alcohol withdrawal. Environmental risks include high heat and humidity, prolonged exertion, immobility, lack of air conditioning, poor acclimatization, and occlusive clothing.
Pediatric considerations Children are at increased risk due to higher body surface area–to–mass ratio and reduced sweating capacity.
Diagnosis
Signs and symptoms Heat stroke presents with the classic triad of hyperthermia, CNS dysfunction, and hot skin. Core temperature exceeds 105°F (40.5°C). Neurologic findings include delirium, coma, seizures, ataxia, and confusion. Cardiovascular findings include tachycardia, hypotension, wide pulse pressure, and conduction abnormalities. Pulmonary findings include tachypnea, respiratory alkalosis, hypoxemia, and noncardiogenic pulmonary edema. GI symptoms include nausea, vomiting, and diarrhea. Renal failure, rhabdomyolysis, hepatic failure with extreme transaminase elevation, and coagulopathy including DIC may occur and indicate poor prognosis. Heat exhaustion causes headache, fatigue, malaise, impaired judgment, nausea, vomiting, tachycardia, dehydration, tachypnea, and profuse sweating without severe CNS dysfunction. Heat cramps cause painful muscle cramps after heavy sweating and hypotonic fluid replacement, leading to hyponatremia. Heat edema presents as dependent lower-extremity swelling in nonacclimatized individuals. Heat syncope presents as transient loss of consciousness during heat exposure, especially in the elderly. Prickly heat causes a pruritic maculopapular or vesicular rash due to sweat duct obstruction.
Essential workup Measure accurate core temperature using a rectal or esophageal probe. Establish history of heat exposure. Heat exhaustion is a diagnosis of exclusion. Heat stroke requires both core temperature >40.5°C and severe CNS dysfunction.
Diagnosis tests and interpretation
Laboratory Obtain CBC (leukocytosis, hemoconcentration), electrolytes, BUN, creatinine, glucose, urinalysis (myoglobin), creatine kinase for rhabdomyolysis, ABG (acidosis and elevated lactate common in exertional heat stroke), blood and urine cultures if sepsis is considered, toxicology screen, PT/PTT and DIC panel, liver function tests, and troponin (elevation suggests poor prognosis).
Imaging ECG in elderly or cardiac risk patients. Chest radiograph for ARDS or aspiration. Head CT for altered mental status. Lumbar puncture may be required to exclude CNS infection.
Differential diagnosis Sepsis, thyroid storm, pheochromocytoma, stimulant or anticholinergic toxicity, meningitis or encephalitis, cerebral malaria, delirium tremens, neuroleptic malignant syndrome, malignant hyperthermia, and serotonin syndrome.
Treatment
Prehospital Remove patient from heat source, disrobe, and initiate cooling with wet sheets.
Initial stabilization/therapy Ensure airway, breathing, and circulation. Begin continuous core temperature monitoring. Initiate rapid cooling if temperature >104°F (40°C). Give IV 0.9% normal saline bolus if hypotensive. For altered mental status, administer glucose, thiamine, and naloxone as indicated.
Emergency department management Cooling is the priority. Use evaporative cooling with misted warm water and high airflow; combine with ice packs to groin and axilla. Cold-water immersion is effective but often impractical. Advanced techniques such as cold peritoneal lavage, extracorporeal circulation, or cold dialysis may be considered for refractory cases. Stop cooling at 102°F (39°C) to prevent hypothermia. Antipyretics are ineffective and alcohol sponge baths should be avoided. Supportive care includes aggressive isotonic fluid resuscitation while avoiding fluid overload. Insert Foley catheter to monitor urine output; target >2 mL/kg/hr if rhabdomyolysis is present. Treat seizures, agitation, and shivering with benzodiazepines. Avoid vasopressors and antiarrhythmics until adequate cooling, as many arrhythmias resolve with temperature normalization. Treat associated heat syndromes with appropriate hydration, electrolyte replacement, elevation or compression, and topical therapy as indicated.
Medication Benzodiazepines such as diazepam or lorazepam for seizures or agitation; naloxone when opioid toxicity is suspected.
Follow-up and disposition
Admission criteria All patients with heat stroke require ICU admission. Heat exhaustion requires admission if severe electrolyte abnormalities, renal failure, rhabdomyolysis, or advanced age are present.
Discharge criteria Patients without heat stroke or severe heat exhaustion who normalize clinically after treatment may be discharged with counseling.
Key points Heat stroke cannot be diagnosed without both core temperature >40.5°C and severe CNS dysfunction. Rapid cooling and supportive care are the cornerstones of management. Continuous core temperature monitoring is standard of care, and evaporative cooling is the preferred first-line method.
