- Published on
Emergency and Acute Medicine – Cauda Equina Syndrome
Core Concept
Cauda equina syndrome results from compression of the lumbar and sacral nerve roots within the cauda equina, which consists of nerve fibers below the conus medullaris, typically ending at the L1–L2 interspace. This condition represents a neurologic emergency because delayed diagnosis and treatment can lead to permanent deficits.
Predisposing Factors
Patients at increased risk include those with neoplastic disease, intravenous drug use, immunocompromised states, and a history of trauma.
Underlying Causes
Lumbar disc herniation is the most common etiology, most frequently involving the L4–L5 level, followed by L5–S1 and L3–L4, and is most often seen in the fourth and fifth decades of life. Other causes include mass lesions such as myeloma, lymphoma, sarcoma, meningioma, neurofibroma, hematoma, and metastatic disease from breast, lung, prostate, thyroid, or renal malignancies. Infectious causes include spinal epidural abscess, particularly in IV drug users. Additional etiologies include blunt or penetrating trauma, complications of spinal anesthesia, and postoperative or spontaneous hematoma.
Clinical Manifestations
Patients commonly report low back pain with unilateral or bilateral radicular symptoms, lower-extremity numbness or weakness, and difficulty ambulating due to pain or motor deficits. Bladder or bowel dysfunction is a key feature and may present as urinary retention, overflow incontinence, or fecal incontinence. On examination, lumbosacral tenderness may be present along with asymmetric lower-extremity sensory or motor deficits, reduced dorsiflexion strength, quadriceps weakness, diminished deep tendon reflexes, saddle hypalgesia or anesthesia, and decreased anal sphincter tone.
Essential Bedside Assessment
A thorough neurologic examination is critical and includes straight-leg raise testing and the Lasègue maneuver, where hip flexion with foot dorsiflexion reproduces posterior thigh pain. Evaluation of perineal sensation, rectal tone, and the anal wink reflex is essential. Measurement of postvoid residual volume by bladder catheterization or ultrasound is strongly recommended, with volumes greater than 50–100 mL considered abnormal, recognizing that residual volume increases with age; a normal residual makes the diagnosis less likely.
Diagnostic Evaluation
Laboratory testing is guided by the suspected differential and may include CBC, urinalysis, ESR, and CRP, particularly when infection or malignancy is a concern. MRI of the spine is the definitive diagnostic study and should be obtained urgently. If MRI is unavailable or contraindicated, CT myelography is an acceptable alternative. Plain radiographs of the lumbosacral spine may be obtained but are insufficient to exclude the diagnosis.
Key Alternative Diagnoses
Conditions that may mimic cauda equina syndrome include lumbosacral strain, osteoarthritis, sciatica, vertebral fractures, osteomyelitis, spinal epidural abscess, conus medullaris or higher spinal cord compression, ankylosing spondylitis, spinal stenosis, abdominal aortic aneurysm or dissection, vascular claudication, hip pathology, and acute transverse myelitis.
Initial Management Priorities
Airway and other traumatic injuries should be managed as indicated. In trauma patients, full spinal immobilization is mandatory, and even in nontrauma presentations, immobilization should be considered because of the possibility of an unstable spinal lesion. Provide adequate analgesia and keep the patient NPO pending neurosurgical evaluation.
Emergency Department Management
Serial neurologic examinations are required to identify progression. Immediate neurosurgical consultation is mandatory in all suspected cases. For acute spinal cord trauma within 8 hours of onset, initiate high-dose methylprednisolone per protocol. If an epidural abscess is suspected, start empiric antibiotics in consultation with neurosurgery. Although there is debate regarding the exact timing of surgical decompression, recommendations generally range from within 6 hours to within 24 hours of symptom onset, with earlier intervention associated with better outcomes.
Pharmacologic Therapy
High-dose methylprednisolone may be administered for acute spinal cord injury using a protocol of a 30 mg/kg IV bolus followed by a continuous infusion of 5.4 mg/kg/hr over 23 hours, provided therapy is initiated within 8 hours of injury.
Disposition and Follow-Up
All patients with acute cauda equina syndrome require hospital admission under neurosurgical care, and treatment should not be delayed. Rapid surgical decompression is associated with improved neurologic recovery, although patients presenting later than 48 hours may still derive benefit. Discharge is appropriate only for patients with a prior complete evaluation, established diagnosis, no new neurologic deficits, and close follow-up arranged with their neurosurgeon.
Clinical Insights and Cautions
Early recognition before irreversible neurologic injury is crucial. Red flags include back pain disproportionate to exam findings, back pain with fever, and back pain in high-risk populations. When infection is suspected, ESR and CRP should be used as screening tools to avoid missed diagnoses.
Core Concept
Cauda equina syndrome results from compression of the lumbar and sacral nerve roots within the cauda equina, which consists of nerve fibers below the conus medullaris, typically ending at the L1–L2 interspace. This condition represents a neurologic emergency because delayed diagnosis and treatment can lead to permanent deficits.
Predisposing Factors
Patients at increased risk include those with neoplastic disease, intravenous drug use, immunocompromised states, and a history of trauma.
Underlying Causes
Lumbar disc herniation is the most common etiology, most frequently involving the L4–L5 level, followed by L5–S1 and L3–L4, and is most often seen in the fourth and fifth decades of life. Other causes include mass lesions such as myeloma, lymphoma, sarcoma, meningioma, neurofibroma, hematoma, and metastatic disease from breast, lung, prostate, thyroid, or renal malignancies. Infectious causes include spinal epidural abscess, particularly in IV drug users. Additional etiologies include blunt or penetrating trauma, complications of spinal anesthesia, and postoperative or spontaneous hematoma.
Clinical Manifestations
Patients commonly report low back pain with unilateral or bilateral radicular symptoms, lower-extremity numbness or weakness, and difficulty ambulating due to pain or motor deficits. Bladder or bowel dysfunction is a key feature and may present as urinary retention, overflow incontinence, or fecal incontinence. On examination, lumbosacral tenderness may be present along with asymmetric lower-extremity sensory or motor deficits, reduced dorsiflexion strength, quadriceps weakness, diminished deep tendon reflexes, saddle hypalgesia or anesthesia, and decreased anal sphincter tone.
Essential Bedside Assessment
A thorough neurologic examination is critical and includes straight-leg raise testing and the Lasègue maneuver, where hip flexion with foot dorsiflexion reproduces posterior thigh pain. Evaluation of perineal sensation, rectal tone, and the anal wink reflex is essential. Measurement of postvoid residual volume by bladder catheterization or ultrasound is strongly recommended, with volumes greater than 50–100 mL considered abnormal, recognizing that residual volume increases with age; a normal residual makes the diagnosis less likely.
Diagnostic Evaluation
Laboratory testing is guided by the suspected differential and may include CBC, urinalysis, ESR, and CRP, particularly when infection or malignancy is a concern. MRI of the spine is the definitive diagnostic study and should be obtained urgently. If MRI is unavailable or contraindicated, CT myelography is an acceptable alternative. Plain radiographs of the lumbosacral spine may be obtained but are insufficient to exclude the diagnosis.
Key Alternative Diagnoses
Conditions that may mimic cauda equina syndrome include lumbosacral strain, osteoarthritis, sciatica, vertebral fractures, osteomyelitis, spinal epidural abscess, conus medullaris or higher spinal cord compression, ankylosing spondylitis, spinal stenosis, abdominal aortic aneurysm or dissection, vascular claudication, hip pathology, and acute transverse myelitis.
Initial Management Priorities
Airway and other traumatic injuries should be managed as indicated. In trauma patients, full spinal immobilization is mandatory, and even in nontrauma presentations, immobilization should be considered because of the possibility of an unstable spinal lesion. Provide adequate analgesia and keep the patient NPO pending neurosurgical evaluation.
Emergency Department Management
Serial neurologic examinations are required to identify progression. Immediate neurosurgical consultation is mandatory in all suspected cases. For acute spinal cord trauma within 8 hours of onset, initiate high-dose methylprednisolone per protocol. If an epidural abscess is suspected, start empiric antibiotics in consultation with neurosurgery. Although there is debate regarding the exact timing of surgical decompression, recommendations generally range from within 6 hours to within 24 hours of symptom onset, with earlier intervention associated with better outcomes.
Pharmacologic Therapy
High-dose methylprednisolone may be administered for acute spinal cord injury using a protocol of a 30 mg/kg IV bolus followed by a continuous infusion of 5.4 mg/kg/hr over 23 hours, provided therapy is initiated within 8 hours of injury.
Disposition and Follow-Up
All patients with acute cauda equina syndrome require hospital admission under neurosurgical care, and treatment should not be delayed. Rapid surgical decompression is associated with improved neurologic recovery, although patients presenting later than 48 hours may still derive benefit. Discharge is appropriate only for patients with a prior complete evaluation, established diagnosis, no new neurologic deficits, and close follow-up arranged with their neurosurgeon.
Clinical Insights and Cautions
Early recognition before irreversible neurologic injury is crucial. Red flags include back pain disproportionate to exam findings, back pain with fever, and back pain in high-risk populations. When infection is suspected, ESR and CRP should be used as screening tools to avoid missed diagnoses.

- Published on
Emergency and Acute Medicine – Carpal Tunnel Syndrome
Condition Overview
Carpal tunnel syndrome results from compression of the median nerve as it traverses the carpal tunnel, a confined space formed by the carpal bones and the transverse carpal ligament. Structures within the tunnel include the median nerve, flexor digitorum profundus, flexor digitorum superficialis, and flexor pollicis longus tendons. The condition may present as an acute or chronic process.
Causes and Predisposing Factors
Acute cases are typically related to trauma, infection, hemorrhage, snake bite, or high-pressure injection injuries. Chronic disease is more common and is associated with repetitive or high-impact occupational activities, pregnancy, oral contraceptive use, granulomatous diseases such as tuberculosis or sarcoidosis, space-occupying lesions causing median nerve compression, osteophytes, amyloid, multiple myeloma, rheumatoid arthritis, endocrine disorders including hypothyroidism, diabetes mellitus, and acromegaly, chronic hemodialysis, or idiopathic causes.
In children, idiopathic disease is rare and most cases are secondary to identifiable causes such as trauma, mucolipidosis, median nerve hamartoma, anomalous flexor digitorum superficialis, or hemophilia with hematoma.
Clinical Presentation
Patients report acute or gradual onset of symptoms. Sensory complaints include numbness and paresthesia in the median nerve distribution involving the thumb, index finger, middle finger, and radial half of the ring finger. Pain is often localized to the wrist or hand but may radiate proximally to the forearm, elbow, or shoulder. Symptoms are classically worse at night and may improve with shaking the hand. Repetitive wrist movements and sustained wrist flexion, such as during driving, often exacerbate symptoms.
On examination, weakness of the abductor pollicis brevis and opponens pollicis may be present, leading to clumsiness, dropping objects, or impaired fine motor control. Loss of two-point discrimination and thenar muscle atrophy are late findings and highly specific.
Key Diagnostic Considerations
Diagnosis is primarily clinical, based on characteristic nocturnal pain and paresthesia in the median nerve distribution. Motor weakness and thenar wasting suggest advanced disease. Provocative maneuvers have limited sensitivity and specificity but may support the diagnosis. These include the Phalen test, Tinel sign at the wrist, carpal compression test with direct pressure over the transverse carpal ligament, and the tourniquet test using a blood pressure cuff.
Investigations and Interpretation
Laboratory studies are generally unnecessary unless systemic disease is suspected; thyroid studies or rheumatologic testing may be considered based on history and examination. Plain wrist radiographs are indicated when trauma or degenerative arthritis is suspected. CT may demonstrate bony encroachment in select cases but is not routine. MRI can visualize soft tissue abnormalities such as median nerve flattening, palmar bowing of the transverse carpal ligament, synovial swelling, or nerve signal changes, though it is not recommended for routine diagnosis. Ultrasound is increasingly used and may show median nerve swelling proximally, distal flattening, and ligament bowing. Nerve conduction studies and electromyography remain the diagnostic gold standard.
Important Alternative Diagnoses
Conditions to consider include cervical radiculopathy involving C6–C7 roots, hand–arm vibration syndrome, thoracic outlet syndrome, first carpometacarpal joint osteoarthritis, brachial plexitis, generalized peripheral neuropathy, syringomyelia, and multiple sclerosis.
Emergency Department Management
No immediate stabilization is typically required. Acute carpal tunnel syndrome constitutes a surgical emergency and warrants urgent hand surgery consultation for decompression of the transverse carpal ligament using open or endoscopic techniques. Chronic disease is managed conservatively with analgesics, activity modification, avoidance of repetitive wrist motion, and wrist splinting in a neutral position, particularly at night. Adjunctive therapies may include yoga and referral for occupational or ergonomic evaluation with tendon and nerve-gliding exercises.
Pharmacologic Therapy
Analgesics may be used for symptom control. NSAIDs have not demonstrated long-term benefit. Short courses of oral corticosteroids provide temporary improvement, such as prednisone 20 mg daily for 7 days followed by 10 mg daily for 7 days, or prednisolone 20–25 mg daily tapered over 2–4 weeks. Local corticosteroid injections offer transient relief in approximately two-thirds of patients and may include hydrocortisone 20 mg, methylprednisolone 15–40 mg, or triamcinolone 20 mg, typically combined with a small volume of lidocaine.
Disposition and Follow-Up
Admission is indicated for acute carpal tunnel syndrome requiring urgent surgical decompression. Patients with chronic disease may be discharged once pain is controlled. Follow-up with a primary care provider, occupational medicine specialist, or hand surgeon should occur within 1–2 weeks for definitive management and consideration of surgical intervention if symptoms persist or progress.
Condition Overview
Carpal tunnel syndrome results from compression of the median nerve as it traverses the carpal tunnel, a confined space formed by the carpal bones and the transverse carpal ligament. Structures within the tunnel include the median nerve, flexor digitorum profundus, flexor digitorum superficialis, and flexor pollicis longus tendons. The condition may present as an acute or chronic process.
Causes and Predisposing Factors
Acute cases are typically related to trauma, infection, hemorrhage, snake bite, or high-pressure injection injuries. Chronic disease is more common and is associated with repetitive or high-impact occupational activities, pregnancy, oral contraceptive use, granulomatous diseases such as tuberculosis or sarcoidosis, space-occupying lesions causing median nerve compression, osteophytes, amyloid, multiple myeloma, rheumatoid arthritis, endocrine disorders including hypothyroidism, diabetes mellitus, and acromegaly, chronic hemodialysis, or idiopathic causes.
In children, idiopathic disease is rare and most cases are secondary to identifiable causes such as trauma, mucolipidosis, median nerve hamartoma, anomalous flexor digitorum superficialis, or hemophilia with hematoma.
Clinical Presentation
Patients report acute or gradual onset of symptoms. Sensory complaints include numbness and paresthesia in the median nerve distribution involving the thumb, index finger, middle finger, and radial half of the ring finger. Pain is often localized to the wrist or hand but may radiate proximally to the forearm, elbow, or shoulder. Symptoms are classically worse at night and may improve with shaking the hand. Repetitive wrist movements and sustained wrist flexion, such as during driving, often exacerbate symptoms.
On examination, weakness of the abductor pollicis brevis and opponens pollicis may be present, leading to clumsiness, dropping objects, or impaired fine motor control. Loss of two-point discrimination and thenar muscle atrophy are late findings and highly specific.
Key Diagnostic Considerations
Diagnosis is primarily clinical, based on characteristic nocturnal pain and paresthesia in the median nerve distribution. Motor weakness and thenar wasting suggest advanced disease. Provocative maneuvers have limited sensitivity and specificity but may support the diagnosis. These include the Phalen test, Tinel sign at the wrist, carpal compression test with direct pressure over the transverse carpal ligament, and the tourniquet test using a blood pressure cuff.
Investigations and Interpretation
Laboratory studies are generally unnecessary unless systemic disease is suspected; thyroid studies or rheumatologic testing may be considered based on history and examination. Plain wrist radiographs are indicated when trauma or degenerative arthritis is suspected. CT may demonstrate bony encroachment in select cases but is not routine. MRI can visualize soft tissue abnormalities such as median nerve flattening, palmar bowing of the transverse carpal ligament, synovial swelling, or nerve signal changes, though it is not recommended for routine diagnosis. Ultrasound is increasingly used and may show median nerve swelling proximally, distal flattening, and ligament bowing. Nerve conduction studies and electromyography remain the diagnostic gold standard.
Important Alternative Diagnoses
Conditions to consider include cervical radiculopathy involving C6–C7 roots, hand–arm vibration syndrome, thoracic outlet syndrome, first carpometacarpal joint osteoarthritis, brachial plexitis, generalized peripheral neuropathy, syringomyelia, and multiple sclerosis.
Emergency Department Management
No immediate stabilization is typically required. Acute carpal tunnel syndrome constitutes a surgical emergency and warrants urgent hand surgery consultation for decompression of the transverse carpal ligament using open or endoscopic techniques. Chronic disease is managed conservatively with analgesics, activity modification, avoidance of repetitive wrist motion, and wrist splinting in a neutral position, particularly at night. Adjunctive therapies may include yoga and referral for occupational or ergonomic evaluation with tendon and nerve-gliding exercises.
Pharmacologic Therapy
Analgesics may be used for symptom control. NSAIDs have not demonstrated long-term benefit. Short courses of oral corticosteroids provide temporary improvement, such as prednisone 20 mg daily for 7 days followed by 10 mg daily for 7 days, or prednisolone 20–25 mg daily tapered over 2–4 weeks. Local corticosteroid injections offer transient relief in approximately two-thirds of patients and may include hydrocortisone 20 mg, methylprednisolone 15–40 mg, or triamcinolone 20 mg, typically combined with a small volume of lidocaine.
Disposition and Follow-Up
Admission is indicated for acute carpal tunnel syndrome requiring urgent surgical decompression. Patients with chronic disease may be discharged once pain is controlled. Follow-up with a primary care provider, occupational medicine specialist, or hand surgeon should occur within 1–2 weeks for definitive management and consideration of surgical intervention if symptoms persist or progress.

- Published on
Emergency and Acute Medicine – Carpal Fractures
Overview
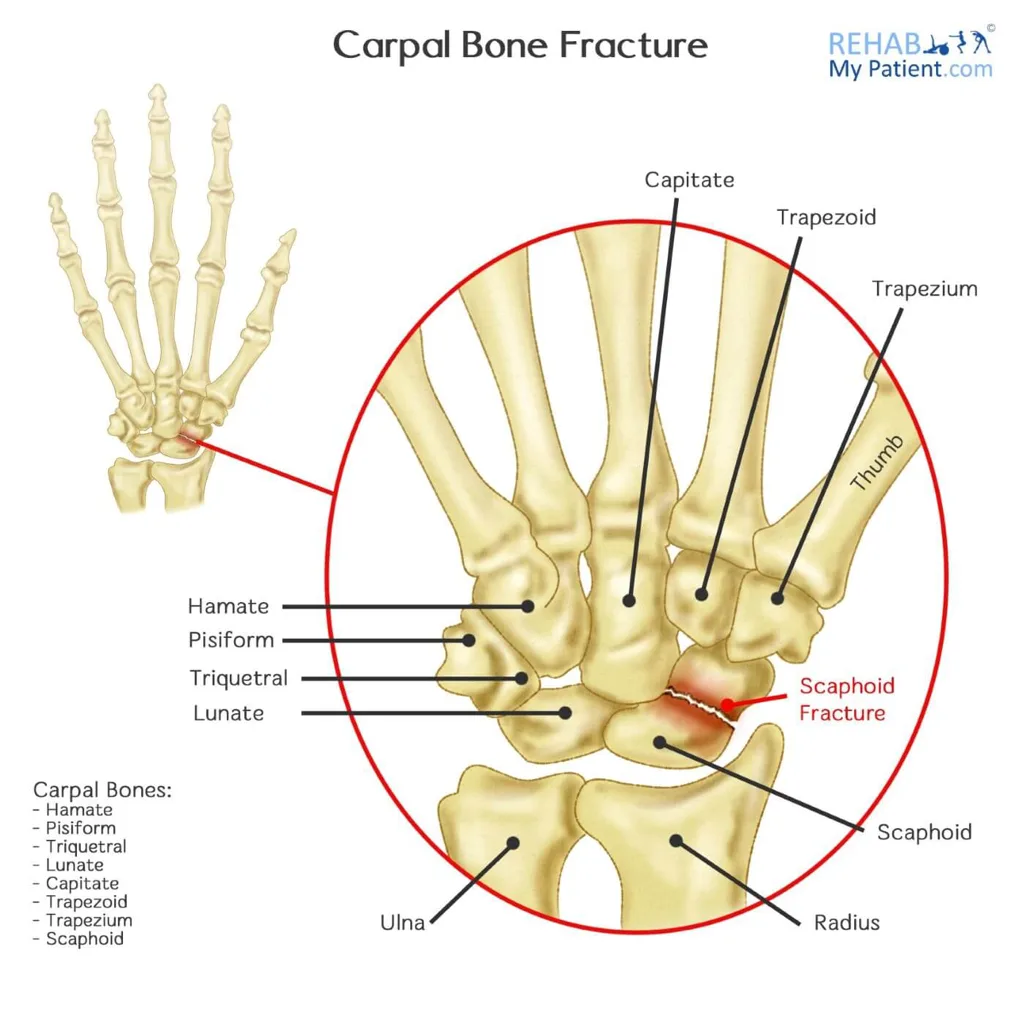
Carpal fractures involve the most frequently injured region of the upper extremity. The scaphoid accounts for approximately 68% of cases, followed by the triquetrum at about 18%. These injuries often coexist with other wrist trauma, including scaphocapitate syndrome (capitate fracture with scaphoid fracture), perilunate dislocations, hamate fractures with fourth and fifth carpometacarpal injuries, and concurrent distal radius fractures.
Mechanisms of Injury
Typical mechanisms include a fall on an outstretched hand with wrist hyperextension or hyperflexion, direct blows, axial loading, and repetitive or chronic use injuries. Hyperflexion commonly results in dorsal avulsion fractures, while hyperextension leads to volar avulsion fractures. Hook of hamate fractures are classically associated with forceful swinging motions, such as with racquets or golf clubs.
Clinical Features
Patients usually report a fall or direct impact. Examination reveals pain, swelling, and reduced wrist motion. Careful palpation of individual carpal bones is possible with proper wrist positioning. Scaphoid fractures often present with snuffbox tenderness, which is sensitive but nonspecific; specificity improves with wrist pronation and ulnar deviation. Axial thumb compression may elicit pain but lacks specificity. Palmar tenderness over the scaphoid tubercle at the distal wrist crease with wrist extension is more specific.
Initial Assessment Priorities
A complete examination of the entire upper extremity and shoulder girdle is required to identify associated injuries. A thorough neurovascular assessment is essential, particularly for hamate fractures, which may involve ulnar nerve or artery injury.
Diagnostic Evaluation
Standard imaging includes anteroposterior, lateral, and oblique views of the wrist and hand. Dedicated views, such as scaphoid views, should be obtained when suspicion remains high. CT scanning provides superior fracture detection, while MRI is useful for identifying occult fractures and ligamentous injuries.
Conditions to Differentiate
Consider metacarpal base fractures, distal radius or ulna fractures, lunate dislocation, perilunate dislocation, and in children, distal radius epiphyseal injuries, as pediatric wrist trauma rarely represents a simple sprain.
Early Management
In the prehospital or early ED setting, protect open wounds, elevate the limb, apply ice, remove constricting jewelry, and immobilize the wrist with padded splints. As with all trauma, assess for life-threatening injuries first.
Definitive Emergency Care
Most isolated carpal fractures are initially treated with immobilization aimed at maintaining alignment. Thumb spica splints are used for scaphoid and trapezium fractures. Sugar-tong splints are appropriate for capitate and lunate fractures, maintaining the wrist in neutral position. Volar splints are suitable for triquetrum, pisiform, trapezoid, and hamate fractures with slight wrist extension. Suspected fractures, especially scaphoid injuries, should be splinted even if initial radiographs are negative. Open fractures require immediate irrigation, IV antibiotics targeting Staphylococcus aureus with additional gram-negative coverage for severe soft tissue injury, tetanus prophylaxis, neurovascular monitoring, and urgent orthopedic consultation.
Pain Control
Analgesia includes NSAIDs, mild oral narcotics, or other oral analgesics. Proper immobilization often provides significant pain relief.
Disposition Planning
Open fractures and unstable or displaced injuries requiring operative intervention warrant admission. Closed, nondisplaced fractures adequately immobilized may be discharged with orthopedic follow-up in 7–10 days.
Follow-Up Strategy
All confirmed fractures require orthopedic referral for casting and definitive care. Missed injuries or inadequate immobilization can result in nonunion, avascular necrosis, chronic pain, and disability. Persistent pain despite negative initial imaging should prompt repeat radiographs or advanced imaging after 7–10 days.
Clinical Pearls and Pitfalls
Carpal fractures are frequently occult on initial imaging and easily missed. All clinically suspected fractures should be immobilized. While most scaphoid fractures occur in isolation, other carpal fractures are commonly associated with additional wrist or hand injuries. Long-term outcomes depend heavily on early recognition, proper splinting in a functional position, and timely orthopedic follow-up.
Overview
Carpal fractures involve the most frequently injured region of the upper extremity. The scaphoid accounts for approximately 68% of cases, followed by the triquetrum at about 18%. These injuries often coexist with other wrist trauma, including scaphocapitate syndrome (capitate fracture with scaphoid fracture), perilunate dislocations, hamate fractures with fourth and fifth carpometacarpal injuries, and concurrent distal radius fractures.
Mechanisms of Injury
Typical mechanisms include a fall on an outstretched hand with wrist hyperextension or hyperflexion, direct blows, axial loading, and repetitive or chronic use injuries. Hyperflexion commonly results in dorsal avulsion fractures, while hyperextension leads to volar avulsion fractures. Hook of hamate fractures are classically associated with forceful swinging motions, such as with racquets or golf clubs.
Clinical Features
Patients usually report a fall or direct impact. Examination reveals pain, swelling, and reduced wrist motion. Careful palpation of individual carpal bones is possible with proper wrist positioning. Scaphoid fractures often present with snuffbox tenderness, which is sensitive but nonspecific; specificity improves with wrist pronation and ulnar deviation. Axial thumb compression may elicit pain but lacks specificity. Palmar tenderness over the scaphoid tubercle at the distal wrist crease with wrist extension is more specific.
Initial Assessment Priorities
A complete examination of the entire upper extremity and shoulder girdle is required to identify associated injuries. A thorough neurovascular assessment is essential, particularly for hamate fractures, which may involve ulnar nerve or artery injury.
Diagnostic Evaluation
Standard imaging includes anteroposterior, lateral, and oblique views of the wrist and hand. Dedicated views, such as scaphoid views, should be obtained when suspicion remains high. CT scanning provides superior fracture detection, while MRI is useful for identifying occult fractures and ligamentous injuries.
Conditions to Differentiate
Consider metacarpal base fractures, distal radius or ulna fractures, lunate dislocation, perilunate dislocation, and in children, distal radius epiphyseal injuries, as pediatric wrist trauma rarely represents a simple sprain.
Early Management
In the prehospital or early ED setting, protect open wounds, elevate the limb, apply ice, remove constricting jewelry, and immobilize the wrist with padded splints. As with all trauma, assess for life-threatening injuries first.
Definitive Emergency Care
Most isolated carpal fractures are initially treated with immobilization aimed at maintaining alignment. Thumb spica splints are used for scaphoid and trapezium fractures. Sugar-tong splints are appropriate for capitate and lunate fractures, maintaining the wrist in neutral position. Volar splints are suitable for triquetrum, pisiform, trapezoid, and hamate fractures with slight wrist extension. Suspected fractures, especially scaphoid injuries, should be splinted even if initial radiographs are negative. Open fractures require immediate irrigation, IV antibiotics targeting Staphylococcus aureus with additional gram-negative coverage for severe soft tissue injury, tetanus prophylaxis, neurovascular monitoring, and urgent orthopedic consultation.
Pain Control
Analgesia includes NSAIDs, mild oral narcotics, or other oral analgesics. Proper immobilization often provides significant pain relief.
Disposition Planning
Open fractures and unstable or displaced injuries requiring operative intervention warrant admission. Closed, nondisplaced fractures adequately immobilized may be discharged with orthopedic follow-up in 7–10 days.
Follow-Up Strategy
All confirmed fractures require orthopedic referral for casting and definitive care. Missed injuries or inadequate immobilization can result in nonunion, avascular necrosis, chronic pain, and disability. Persistent pain despite negative initial imaging should prompt repeat radiographs or advanced imaging after 7–10 days.
Clinical Pearls and Pitfalls
Carpal fractures are frequently occult on initial imaging and easily missed. All clinically suspected fractures should be immobilized. While most scaphoid fractures occur in isolation, other carpal fractures are commonly associated with additional wrist or hand injuries. Long-term outcomes depend heavily on early recognition, proper splinting in a functional position, and timely orthopedic follow-up.
- Published on
Emergency and Acute Medicine – Peripartum Cardiomyopathy
Core Definition
Peripartum cardiomyopathy is a dilated cardiomyopathy that develops during the final month of pregnancy or within the first 5 months after delivery. Diagnosis requires all of the following: onset of heart failure in the defined peripartum period, no identifiable alternative cause, no preexisting cardiac disease, and objective echocardiographic evidence of systolic dysfunction. Incidence is approximately 3–5 per 10,000 live births. About half of cases recover spontaneously. Reported mortality ranges from 18% to 56%. Thromboembolic complications are more common than in other cardiomyopathies.
Risk Profile and Prognostic Indicators
Risk is increased in women older than 30 years, multiparity, multiple gestations, prolonged tocolytic therapy exceeding 4 weeks, obesity, preeclampsia, and African American race. Poor prognosis is associated with low left ventricular ejection fraction at 6 months postpartum, symptom onset more than 2 weeks after delivery, age over 30 years, African American descent, and multiparity.
Proposed Pathophysiology
The exact cause remains uncertain. Leading theories include viral myocarditis facilitated by pregnancy-related immunosuppression, autoimmune reaction to maternal or fetal antigens, maladaptive response to pregnancy-related hemodynamic stress, stress-induced cytokine activation, prolonged tocolysis, and nutritional deficiencies such as selenium deficiency.
Clinical Presentation
Common symptoms include dyspnea, orthopnea, paroxysmal nocturnal dyspnea, cough, dizziness, chest discomfort, fatigue, anorexia, and arrhythmias. History should focus on timing of symptom onset, persistent unexplained cough, excessive weight gain greater than 2–4 lb per week, prior cardiac disease, and complications in previous pregnancies. Examination findings may include jugular venous distention, gallop rhythm, mitral regurgitation murmur, loud P2, pulmonary rales, rapid-onset peripheral edema, hepatomegaly, and hepatojugular reflux.
Essential Evaluation
Chest radiography may show pulmonary venous congestion, cardiomegaly (difficult to interpret in pregnancy), and pleural effusions. Electrocardiography is nonspecific but may demonstrate left ventricular hypertrophy, left atrial enlargement, T-wave changes, ventricular ectopy (≈40%), or atrial fibrillation (≈20%).
Diagnostic Data Interpretation
Laboratory studies typically show normal electrolytes and renal function. Mild postpartum anemia may worsen dyspnea and fatigue. BNP is useful in differentiating cardiac from pulmonary causes of dyspnea; levels >100 pg/mL strongly support heart failure, while ≤50 pg/mL has a high negative predictive value. Chest imaging may demonstrate progressive pulmonary edema patterns from cephalization to interstitial and alveolar edema, sometimes mimicking pneumonia. Echocardiography confirms diagnosis by showing global ventricular dilation, wall thinning, and reduced systolic function. Diagnostic criteria include ejection fraction <45%, fractional shortening <30%, and indexed lv end-diastolic dimension>2.72 cm/m², with exclusion of valvular disease and tamponade. Endomyocardial biopsy is reserved for suspected myocarditis or consideration of immunosuppressive therapy.
Conditions to Exclude
Alternative causes of dilated cardiomyopathy include ischemia, infarction, valvular disease, chronic hypertension, familial cardiomyopathy, alcohol or drug toxicity, metabolic disorders, thyroid disease, infections, autoimmune disease, neuromuscular disorders, and mitochondrial disease. Other causes of dyspnea or edema include pulmonary embolism, pneumonia, asthma, anemia, hyperthyroidism, constrictive pericarditis, tamponade, nephrotic syndrome, and cirrhosis.
Initial and Emergency Management
Differentiate pulmonary edema from reactive airway disease early. Rapid assessment of airway, breathing, and circulation is essential. Provide supplemental oxygen, consider noninvasive ventilation, and address preload and afterload carefully.
Antepartum management includes nitrates, hydralazine, IV diuretics, amlodipine, digoxin for rate control, cautious use of carvedilol when stable, low-molecular-weight heparin when EF <35%, fetal monitoring, and invasive monitoring if unstable.
Postpartum therapy allows addition of ACE inhibitors or ARBs. Anticoagulation is frequently recommended due to high thromboembolic risk. Heparin is preferred during pregnancy; warfarin is avoided until postpartum. Refractory or severe cases may require inotropes, vasopressors, vasodilators, mechanical circulatory support, or extracorporeal membrane oxygenation. Immunosuppressive therapy may be considered if no improvement occurs after 2 weeks of optimal medical therapy, though evidence remains mixed.
Disposition Decisions
All symptomatic patients with new-onset peripartum cardiomyopathy require admission. ICU care is indicated for pulmonary edema, cardiogenic shock, or ischemia. Discharge may be considered only in carefully selected patients with mild dysfunction, established disease, rapid symptom resolution, no ischemia, and reliable close follow-up.
Follow-Up Guidance
Cardiology follow-up is mandatory. Patients should maintain adequate hydration, restrict dietary sodium, avoid alcohol, use compression stockings if needed, monitor daily weights, and seek care for rapid weight gain, worsening dyspnea, syncope, or palpitations.
Clinical Pearls and Pitfalls
Thromboembolism risk is high in pregnancy-associated cardiomyopathy. Early recognition and coordinated management with cardiology and obstetrics significantly improve outcomes.
Core Definition
Peripartum cardiomyopathy is a dilated cardiomyopathy that develops during the final month of pregnancy or within the first 5 months after delivery. Diagnosis requires all of the following: onset of heart failure in the defined peripartum period, no identifiable alternative cause, no preexisting cardiac disease, and objective echocardiographic evidence of systolic dysfunction. Incidence is approximately 3–5 per 10,000 live births. About half of cases recover spontaneously. Reported mortality ranges from 18% to 56%. Thromboembolic complications are more common than in other cardiomyopathies.
Risk Profile and Prognostic Indicators
Risk is increased in women older than 30 years, multiparity, multiple gestations, prolonged tocolytic therapy exceeding 4 weeks, obesity, preeclampsia, and African American race. Poor prognosis is associated with low left ventricular ejection fraction at 6 months postpartum, symptom onset more than 2 weeks after delivery, age over 30 years, African American descent, and multiparity.
Proposed Pathophysiology
The exact cause remains uncertain. Leading theories include viral myocarditis facilitated by pregnancy-related immunosuppression, autoimmune reaction to maternal or fetal antigens, maladaptive response to pregnancy-related hemodynamic stress, stress-induced cytokine activation, prolonged tocolysis, and nutritional deficiencies such as selenium deficiency.
Clinical Presentation
Common symptoms include dyspnea, orthopnea, paroxysmal nocturnal dyspnea, cough, dizziness, chest discomfort, fatigue, anorexia, and arrhythmias. History should focus on timing of symptom onset, persistent unexplained cough, excessive weight gain greater than 2–4 lb per week, prior cardiac disease, and complications in previous pregnancies. Examination findings may include jugular venous distention, gallop rhythm, mitral regurgitation murmur, loud P2, pulmonary rales, rapid-onset peripheral edema, hepatomegaly, and hepatojugular reflux.
Essential Evaluation
Chest radiography may show pulmonary venous congestion, cardiomegaly (difficult to interpret in pregnancy), and pleural effusions. Electrocardiography is nonspecific but may demonstrate left ventricular hypertrophy, left atrial enlargement, T-wave changes, ventricular ectopy (≈40%), or atrial fibrillation (≈20%).
Diagnostic Data Interpretation
Laboratory studies typically show normal electrolytes and renal function. Mild postpartum anemia may worsen dyspnea and fatigue. BNP is useful in differentiating cardiac from pulmonary causes of dyspnea; levels >100 pg/mL strongly support heart failure, while ≤50 pg/mL has a high negative predictive value. Chest imaging may demonstrate progressive pulmonary edema patterns from cephalization to interstitial and alveolar edema, sometimes mimicking pneumonia. Echocardiography confirms diagnosis by showing global ventricular dilation, wall thinning, and reduced systolic function. Diagnostic criteria include ejection fraction <45%, fractional shortening <30%, and indexed lv end-diastolic dimension>2.72 cm/m², with exclusion of valvular disease and tamponade. Endomyocardial biopsy is reserved for suspected myocarditis or consideration of immunosuppressive therapy.
Conditions to Exclude
Alternative causes of dilated cardiomyopathy include ischemia, infarction, valvular disease, chronic hypertension, familial cardiomyopathy, alcohol or drug toxicity, metabolic disorders, thyroid disease, infections, autoimmune disease, neuromuscular disorders, and mitochondrial disease. Other causes of dyspnea or edema include pulmonary embolism, pneumonia, asthma, anemia, hyperthyroidism, constrictive pericarditis, tamponade, nephrotic syndrome, and cirrhosis.
Initial and Emergency Management
Differentiate pulmonary edema from reactive airway disease early. Rapid assessment of airway, breathing, and circulation is essential. Provide supplemental oxygen, consider noninvasive ventilation, and address preload and afterload carefully.
Antepartum management includes nitrates, hydralazine, IV diuretics, amlodipine, digoxin for rate control, cautious use of carvedilol when stable, low-molecular-weight heparin when EF <35%, fetal monitoring, and invasive monitoring if unstable.
Postpartum therapy allows addition of ACE inhibitors or ARBs. Anticoagulation is frequently recommended due to high thromboembolic risk. Heparin is preferred during pregnancy; warfarin is avoided until postpartum. Refractory or severe cases may require inotropes, vasopressors, vasodilators, mechanical circulatory support, or extracorporeal membrane oxygenation. Immunosuppressive therapy may be considered if no improvement occurs after 2 weeks of optimal medical therapy, though evidence remains mixed.
Disposition Decisions
All symptomatic patients with new-onset peripartum cardiomyopathy require admission. ICU care is indicated for pulmonary edema, cardiogenic shock, or ischemia. Discharge may be considered only in carefully selected patients with mild dysfunction, established disease, rapid symptom resolution, no ischemia, and reliable close follow-up.
Follow-Up Guidance
Cardiology follow-up is mandatory. Patients should maintain adequate hydration, restrict dietary sodium, avoid alcohol, use compression stockings if needed, monitor daily weights, and seek care for rapid weight gain, worsening dyspnea, syncope, or palpitations.
Clinical Pearls and Pitfalls
Thromboembolism risk is high in pregnancy-associated cardiomyopathy. Early recognition and coordinated management with cardiology and obstetrics significantly improve outcomes.

- Published on
Emergency and Acute Medicine – Hypertrophic Cardiomyopathy
Core Overview
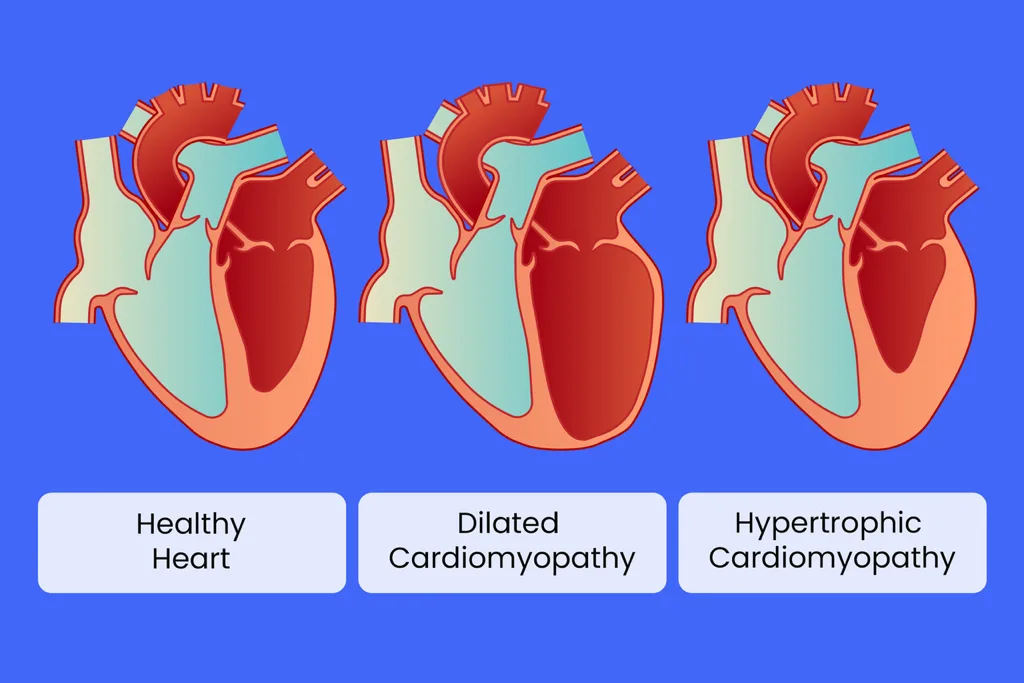
Hypertrophic cardiomyopathy (HCM) is a genetic disorder of the cardiac sarcomere caused by numerous mutations, resulting in a hypertrophied, nondilated left ventricle (and rarely right ventricle) without another cause such as hypertension or aortic stenosis. Phenotypic expression is highly variable. Two major forms exist: nonobstructive HCM, accounting for approximately 75% of patients with an estimated annual mortality of ~1%, and obstructive hypertrophic cardiomyopathy (HOCM), accounting for ~25% with more severe disease and an estimated annual mortality of ~2%.
HCM can present at any age, from neonates to the elderly, but most cases manifest during childhood or adolescence, often coinciding with the pubertal growth spurt. Disease diagnosed at younger ages is typically more severe. A small subset progresses to reduced left ventricular systolic function later in life. Adult-onset presentations are not uncommon and are frequently misdiagnosed as asthma, COPD, deconditioning, or sleep apnea. Lethal ventricular arrhythmias are more common in younger patients, while supraventricular arrhythmias increase with age. Atrial fibrillation is common and often poorly tolerated. HCM is the most common cause of atraumatic sudden death in young athletes under 35 years of age. Prevalence is approximately 1 in 500 adults based on echocardiographic screening.
Structural and Pathophysiologic Features
Pathology is characterized by marked and irregular ventricular wall thickening with myofibrillar disarray and fibrin deposition, predominantly affecting the high-pressure left ventricle. Septal hypertrophy is usually more pronounced than free wall thickening. In obstructive disease, hypertrophy may regress if outflow obstruction is relieved. Some phenotypes demonstrate progressive wall thinning with age, typically following marked hypertrophy earlier in life. Diastolic stiffness leads to atrial dilation. Microvascular dysfunction occurs due to impaired vasodilation, intimal thickening, and perivascular collagen deposition.
Long-Term Outpatient Management Principles
Management focuses on avoiding volume depletion and excessive cardiac demand, with therapy tailored to hypertrophy severity and distribution. Pharmacologic treatment includes beta-blockers or verapamil to slow heart rate and prolong diastole. Implantable cardioverter-defibrillators are indicated in patients with prior syncope, cardiac arrest, family history of sudden death, nonsustained ventricular tachycardia, abnormal blood pressure response to exercise, or massive hypertrophy. Septal reduction strategies include alcohol septal ablation and surgical septal myectomy, with improving outcomes at experienced centers.
Genetic Risk Profile
HCM was the first cardiac disease with an identified genetic basis (1989). It follows an autosomal-dominant inheritance pattern with more than 10 associated genes, most encoding sarcomeric proteins. Over 700 distinct mutations have been identified, with high penetrance and marked phenotypic variability. Some genotypes carry significantly higher risk of sudden death. Routine genotype-based risk stratification remains impractical due to complexity and incomplete understanding of modifier genes and environmental factors. Certain mutations involving cell membrane pumps are associated with increased arrhythmogenic risk.
Clinical Presentation
Symptoms often correlate with exertion or sudden upright posture, both of which reduce venous return and ventricular filling. Severity depends on hypertrophy location and extent. Common symptoms include exertional dyspnea, shortness of breath, postprandial or exertional angina, presyncope, syncope, congestive heart failure, cardiovascular collapse, and dysrhythmias. Paroxysmal atrial fibrillation may precipitate rapid deterioration, particularly in heart failure, and increases thromboembolic risk. Ventricular tachycardia or fibrillation may cause sudden death, while bradyarrhythmias are rare.
Patients may have a history of septal myectomy or alcohol ablation, with associated risks of conduction abnormalities or septal rupture. High-risk individuals may have implanted defibrillators. A family history of unexplained sudden death or known HCM is a critical diagnostic clue.
Pediatric Considerations
Disease severity may increase during adolescence. Any child with unexplained syncope, especially during exertion, warrants detailed family history (standard three-generation pedigree) and specialist referral.
Physical Examination Findings
Findings may be absent or subtle in nonobstructive disease. In obstructive HCM, findings may include a loud left-sided S4, a double apical impulse, and a crescendo–decrescendo midsystolic murmur best heard at the left sternal border. The murmur increases with standing or Valsalva and decreases with squatting, recumbency, or handgrip. Mitral regurgitation is common; radiation to the axilla suggests associated mitral insufficiency.
Electrocardiographic Features
ECG is abnormal in over 90% of patients. Findings include T-wave inversion >1 mm in multiple leads, ST-segment depression >0.5 mm, deep Q waves (>3 mm or >40 ms) excluding leads III and aVR, prolonged P waves with negative terminal forces in V1, and nonspecific intraventricular conduction delay >140 ms. Athletic ECG variants require careful interpretation, especially in non–African-Caribbean athletes over 16 years.
Laboratory and Imaging Evaluation
Routine laboratory testing has no diagnostic value in the emergency setting. Genetic testing is outpatient-focused. Chest radiography is usually normal but may show left ventricular free wall bulging, atrial enlargement, or pulmonary vascular redistribution. Transthoracic echocardiography with Doppler is the diagnostic cornerstone, demonstrating LV wall thickness >15 mm in adults (or ≥13–14 mm with supportive features), small or normal LV cavity, systolic outflow obstruction, and diastolic dysfunction. In children, wall thickness ≥2 standard deviations above the mean is diagnostic. Cardiac MRI supplements echocardiography, offering detailed assessment of hypertrophy distribution and fibrosis. Stress thallium and PET imaging may identify ischemia.
Differential Diagnosis
Consider vasovagal syncope, heat illness, aortic or pulmonic stenosis, ventricular septal defect, mitral regurgitation or prolapse, and coronary artery disease. Angina or heart failure in HCM carries higher risk than in non-HCM populations.
Emergency Management Principles
Suspect HCM in patients who deteriorate with standard heart failure, ischemia, or supraventricular tachycardia treatment, and in young athletes collapsing during exertion. Patients are highly sensitive to preload reduction and impaired diastolic filling. Initial management includes airway stabilization, oxygen, IV access, cardiac monitoring, and pulse oximetry. Patients may need to remain supine.
Avoid routine vasodilators for CHF or angina, as they may precipitate collapse. If hypotension occurs, administer cautious fluid boluses. Even mild hypovolemia can significantly reduce cardiac output. Rate control and improved diastolic filling are central goals. Beta-blockers are first-line therapy; verapamil is useful but nifedipine is relatively contraindicated due to vasodilation.
Supraventricular dysrhythmias are treated initially with beta-blockers or calcium channel blockers. Amiodarone is the drug of choice for ventricular dysrhythmias or refractory cases. Early electrical cardioversion is recommended for new-onset atrial fibrillation with heart failure.
Key Medications
Amiodarone, propranolol, verapamil, phenylephrine (for refractory hypotension without inotropic effect), and diltiazem (contraindicated in children under 12) may be used with careful hemodynamic assessment. All medications must be evaluated for their impact on outflow tract obstruction.
Disposition Decisions
Admission is required for unexplained syncope, heart failure, angina, or hemodynamically significant tachydysrhythmias, often to ICU level care. Discharge may be considered only when hypertrophy is an incidental finding without personal or family history of syncope or sudden death, with urgent cardiology follow-up arranged. Patients must be counseled to avoid exertion or situations that reduce preload until evaluated.
Clinical Pitfalls and Key Insights
Genetic and phenotypic diversity complicates diagnosis and risk stratification. Some advocate routine echocardiographic screening for youth sports participation or ICD placement at diagnosis. Any patient with syncope in whom HCM is suspected must be fully evaluated, as recurrence carries a high risk of sudden death.
Core Overview
Hypertrophic cardiomyopathy (HCM) is a genetic disorder of the cardiac sarcomere caused by numerous mutations, resulting in a hypertrophied, nondilated left ventricle (and rarely right ventricle) without another cause such as hypertension or aortic stenosis. Phenotypic expression is highly variable. Two major forms exist: nonobstructive HCM, accounting for approximately 75% of patients with an estimated annual mortality of ~1%, and obstructive hypertrophic cardiomyopathy (HOCM), accounting for ~25% with more severe disease and an estimated annual mortality of ~2%.
HCM can present at any age, from neonates to the elderly, but most cases manifest during childhood or adolescence, often coinciding with the pubertal growth spurt. Disease diagnosed at younger ages is typically more severe. A small subset progresses to reduced left ventricular systolic function later in life. Adult-onset presentations are not uncommon and are frequently misdiagnosed as asthma, COPD, deconditioning, or sleep apnea. Lethal ventricular arrhythmias are more common in younger patients, while supraventricular arrhythmias increase with age. Atrial fibrillation is common and often poorly tolerated. HCM is the most common cause of atraumatic sudden death in young athletes under 35 years of age. Prevalence is approximately 1 in 500 adults based on echocardiographic screening.
Structural and Pathophysiologic Features
Pathology is characterized by marked and irregular ventricular wall thickening with myofibrillar disarray and fibrin deposition, predominantly affecting the high-pressure left ventricle. Septal hypertrophy is usually more pronounced than free wall thickening. In obstructive disease, hypertrophy may regress if outflow obstruction is relieved. Some phenotypes demonstrate progressive wall thinning with age, typically following marked hypertrophy earlier in life. Diastolic stiffness leads to atrial dilation. Microvascular dysfunction occurs due to impaired vasodilation, intimal thickening, and perivascular collagen deposition.
Long-Term Outpatient Management Principles
Management focuses on avoiding volume depletion and excessive cardiac demand, with therapy tailored to hypertrophy severity and distribution. Pharmacologic treatment includes beta-blockers or verapamil to slow heart rate and prolong diastole. Implantable cardioverter-defibrillators are indicated in patients with prior syncope, cardiac arrest, family history of sudden death, nonsustained ventricular tachycardia, abnormal blood pressure response to exercise, or massive hypertrophy. Septal reduction strategies include alcohol septal ablation and surgical septal myectomy, with improving outcomes at experienced centers.
Genetic Risk Profile
HCM was the first cardiac disease with an identified genetic basis (1989). It follows an autosomal-dominant inheritance pattern with more than 10 associated genes, most encoding sarcomeric proteins. Over 700 distinct mutations have been identified, with high penetrance and marked phenotypic variability. Some genotypes carry significantly higher risk of sudden death. Routine genotype-based risk stratification remains impractical due to complexity and incomplete understanding of modifier genes and environmental factors. Certain mutations involving cell membrane pumps are associated with increased arrhythmogenic risk.
Clinical Presentation
Symptoms often correlate with exertion or sudden upright posture, both of which reduce venous return and ventricular filling. Severity depends on hypertrophy location and extent. Common symptoms include exertional dyspnea, shortness of breath, postprandial or exertional angina, presyncope, syncope, congestive heart failure, cardiovascular collapse, and dysrhythmias. Paroxysmal atrial fibrillation may precipitate rapid deterioration, particularly in heart failure, and increases thromboembolic risk. Ventricular tachycardia or fibrillation may cause sudden death, while bradyarrhythmias are rare.
Patients may have a history of septal myectomy or alcohol ablation, with associated risks of conduction abnormalities or septal rupture. High-risk individuals may have implanted defibrillators. A family history of unexplained sudden death or known HCM is a critical diagnostic clue.
Pediatric Considerations
Disease severity may increase during adolescence. Any child with unexplained syncope, especially during exertion, warrants detailed family history (standard three-generation pedigree) and specialist referral.
Physical Examination Findings
Findings may be absent or subtle in nonobstructive disease. In obstructive HCM, findings may include a loud left-sided S4, a double apical impulse, and a crescendo–decrescendo midsystolic murmur best heard at the left sternal border. The murmur increases with standing or Valsalva and decreases with squatting, recumbency, or handgrip. Mitral regurgitation is common; radiation to the axilla suggests associated mitral insufficiency.
Electrocardiographic Features
ECG is abnormal in over 90% of patients. Findings include T-wave inversion >1 mm in multiple leads, ST-segment depression >0.5 mm, deep Q waves (>3 mm or >40 ms) excluding leads III and aVR, prolonged P waves with negative terminal forces in V1, and nonspecific intraventricular conduction delay >140 ms. Athletic ECG variants require careful interpretation, especially in non–African-Caribbean athletes over 16 years.
Laboratory and Imaging Evaluation
Routine laboratory testing has no diagnostic value in the emergency setting. Genetic testing is outpatient-focused. Chest radiography is usually normal but may show left ventricular free wall bulging, atrial enlargement, or pulmonary vascular redistribution. Transthoracic echocardiography with Doppler is the diagnostic cornerstone, demonstrating LV wall thickness >15 mm in adults (or ≥13–14 mm with supportive features), small or normal LV cavity, systolic outflow obstruction, and diastolic dysfunction. In children, wall thickness ≥2 standard deviations above the mean is diagnostic. Cardiac MRI supplements echocardiography, offering detailed assessment of hypertrophy distribution and fibrosis. Stress thallium and PET imaging may identify ischemia.
Differential Diagnosis
Consider vasovagal syncope, heat illness, aortic or pulmonic stenosis, ventricular septal defect, mitral regurgitation or prolapse, and coronary artery disease. Angina or heart failure in HCM carries higher risk than in non-HCM populations.
Emergency Management Principles
Suspect HCM in patients who deteriorate with standard heart failure, ischemia, or supraventricular tachycardia treatment, and in young athletes collapsing during exertion. Patients are highly sensitive to preload reduction and impaired diastolic filling. Initial management includes airway stabilization, oxygen, IV access, cardiac monitoring, and pulse oximetry. Patients may need to remain supine.
Avoid routine vasodilators for CHF or angina, as they may precipitate collapse. If hypotension occurs, administer cautious fluid boluses. Even mild hypovolemia can significantly reduce cardiac output. Rate control and improved diastolic filling are central goals. Beta-blockers are first-line therapy; verapamil is useful but nifedipine is relatively contraindicated due to vasodilation.
Supraventricular dysrhythmias are treated initially with beta-blockers or calcium channel blockers. Amiodarone is the drug of choice for ventricular dysrhythmias or refractory cases. Early electrical cardioversion is recommended for new-onset atrial fibrillation with heart failure.
Key Medications
Amiodarone, propranolol, verapamil, phenylephrine (for refractory hypotension without inotropic effect), and diltiazem (contraindicated in children under 12) may be used with careful hemodynamic assessment. All medications must be evaluated for their impact on outflow tract obstruction.
Disposition Decisions
Admission is required for unexplained syncope, heart failure, angina, or hemodynamically significant tachydysrhythmias, often to ICU level care. Discharge may be considered only when hypertrophy is an incidental finding without personal or family history of syncope or sudden death, with urgent cardiology follow-up arranged. Patients must be counseled to avoid exertion or situations that reduce preload until evaluated.
Clinical Pitfalls and Key Insights
Genetic and phenotypic diversity complicates diagnosis and risk stratification. Some advocate routine echocardiographic screening for youth sports participation or ICD placement at diagnosis. Any patient with syncope in whom HCM is suspected must be fully evaluated, as recurrence carries a high risk of sudden death.
- Published on
Emergency and Acute Medicine – Cardiomyopathy
Foundational Overview
Cardiomyopathies are disorders of the myocardium associated with structural and functional cardiac impairment. Major forms include dilated cardiomyopathy, hypertrophic cardiomyopathy, restrictive cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, and unclassified variants. Specific cardiomyopathies occur secondary to systemic diseases or identifiable conditions. Dilated cardiomyopathy accounts for approximately 25% of all heart failure cases and is the most common subtype encountered in emergency care.
Causative Mechanisms
Dilated cardiomyopathy may be idiopathic, viral, genetic, toxic, immune mediated, or familial. Hypertrophic cardiomyopathy is typically an autosomal dominant inherited disorder. Restrictive cardiomyopathy may be idiopathic or related to infiltrative diseases such as amyloidosis. Arrhythmogenic right ventricular cardiomyopathy is usually familial with either dominant or recessive inheritance. Secondary cardiomyopathies include infectious causes such as viral myocarditis, Lyme disease, Chagas disease, and HIV; toxic causes such as alcohol, chemotherapeutic agents, and peripartum states; metabolic causes including hyperthyroidism, pheochromocytoma, and stress-induced (Takotsubo) cardiomyopathy; and systemic diseases such as lupus, scleroderma, neuromuscular disorders, and amyloidosis.
Pediatric-Specific Etiologies
In children, cardiomyopathy may be idiopathic or genetic, including inborn errors of metabolism, neuromuscular disease, malformation syndromes, and familial isolated cardiomyopathies. Acquired causes include nutritional deficiencies, electrolyte and endocrine disturbances, toxins, collagen vascular disease, immunologic disorders, malignancy, morbid obesity, myocarditis, pulmonary disease, Kawasaki disease, infections, radiation, congenital heart disease, and perinatal asphyxia.
Clinical Manifestations
History may reveal antecedent viral illness, chemotherapy exposure, HIV, Lyme disease, pregnancy, substance use, or systemic conditions such as hemochromatosis or sarcoidosis. Family history of sudden cardiac death is a critical clue. Symptoms include exertional dyspnea, dizziness, palpitations, near-syncope or syncope, ventricular arrhythmias, and congestive heart failure. Pediatric presentations may include irritability, hepatomegaly, generalized weakness, hypoglycemia, metabolic acidosis, hyperammonemia, cyanosis, encephalopathy, and dysmorphic features. Pregnancy-related cardiomyopathy should be considered separately.
Initial Assessment Priorities
Evaluation focuses on vital signs, cardiopulmonary examination, evidence of volume overload, abdominal organomegaly, peripheral edema, and signs of systemic disease such as rash or goiter. Rapid identification of decompensated heart failure, malignant arrhythmias, or shock is essential.
Diagnostic Evaluation
Laboratory studies include CBC, metabolic panel, liver and thyroid function tests, cardiac biomarkers, and BNP (levels >100 pg/mL support heart failure). Serologic testing is rarely helpful in the emergency setting. Chest radiography may demonstrate cardiomegaly, pulmonary congestion, and pleural effusions in dilated cardiomyopathy, whereas restrictive cardiomyopathy often shows a normal cardiac silhouette with pulmonary congestion. Emergency bedside transthoracic echocardiography may reveal depressed LV ejection fraction and exclude pericardial tamponade. Formal echocardiography is the diagnostic study of choice to characterize chamber size, wall thickness, systolic and diastolic function, and valvular disease. CT and cardiac MRI help differentiate restrictive cardiomyopathy from constrictive pericarditis and provide detailed assessment of myocardial anatomy, fibrosis, infiltration, iron overload, and viability.
Electrocardiographic Patterns
Hypertrophic cardiomyopathy commonly shows LV hypertrophy and deep Q waves in inferolateral leads, particularly in adolescents. Dilated, toxic, Lyme, and Chagas cardiomyopathies may present with atrial fibrillation, heart block, conduction delays, or pseudoinfarct patterns. Stress-induced (Takotsubo) cardiomyopathy may mimic STEMI and often necessitates cardiac catheterization to exclude ischemia.
Important Differentials
Other causes of dyspnea include COPD, asthma, anemia, interstitial lung disease, pulmonary embolism, pericardial tamponade, valvular disease, ischemic heart disease, hypothyroidism, and constrictive pericarditis. Syncope differentials include hypovolemia, hypoglycemia, heat illness, arrhythmia, and cardiac ischemia.
Prehospital Considerations
Patients require monitoring, supplemental oxygen, and cautious use of nitrates, particularly in suspected hypertrophic cardiomyopathy. Decompensated heart failure may benefit from nitrates and noninvasive positive-pressure ventilation.
Emergency Stabilization
Management prioritizes airway, breathing, and circulation with oxygen supplementation and noninvasive ventilation when indicated. Intubation is reserved for respiratory failure or severe encephalopathy.
Definitive Emergency Management
Treatment focuses on standard heart failure and arrhythmia protocols. Anticoagulation is indicated in dilated cardiomyopathy with atrial fibrillation or embolic risk. Atrial fibrillation and ventricular dysrhythmias are managed per ACLS principles. In hypertrophic cardiomyopathy, negative inotropes such as beta-blockers or disopyramide may reduce outflow obstruction. Special pediatric considerations include keeping patients NPO until inborn metabolic errors are excluded, administering dextrose-containing fluids cautiously, and avoiding lactate-containing solutions. Carnitine, antioxidants, and vitamin cofactors may be required in metabolic disorders.
Common Emergency Medications
Therapies may include amiodarone, beta-blockers, calcium channel blockers, diuretics, nitrates, anticoagulation, milrinone, nesiritide, and disease-specific agents such as carnitine, with careful age- and comorbidity-adjusted dosing.
Disposition Planning
Admission is required for new or suspected cardiomyopathy, syncope with concern for arrhythmia or hypertrophic cardiomyopathy, family history of sudden death, cardiogenic shock, or significant decompensation. Discharge may be considered for known cardiomyopathy with mild heart failure responding to treatment, typically after cardiology consultation.
Referral and Follow-Up
Patients with reduced ejection fraction (<35%) may require referral for implantable cardioverter-defibrillators, biventricular pacing, ventricular assist devices, or transplant evaluation. ongoing cardiology follow-up and consideration of genetic testing are essential.< />pan>
Clinical Insights
Bedside echocardiography is a powerful emergency tool in patients with syncope or exertional symptoms. A thorough family history is critical, as inherited cardiomyopathies are a major cause of sudden cardiac death in young patients.
Foundational Overview
Cardiomyopathies are disorders of the myocardium associated with structural and functional cardiac impairment. Major forms include dilated cardiomyopathy, hypertrophic cardiomyopathy, restrictive cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, and unclassified variants. Specific cardiomyopathies occur secondary to systemic diseases or identifiable conditions. Dilated cardiomyopathy accounts for approximately 25% of all heart failure cases and is the most common subtype encountered in emergency care.
Causative Mechanisms
Dilated cardiomyopathy may be idiopathic, viral, genetic, toxic, immune mediated, or familial. Hypertrophic cardiomyopathy is typically an autosomal dominant inherited disorder. Restrictive cardiomyopathy may be idiopathic or related to infiltrative diseases such as amyloidosis. Arrhythmogenic right ventricular cardiomyopathy is usually familial with either dominant or recessive inheritance. Secondary cardiomyopathies include infectious causes such as viral myocarditis, Lyme disease, Chagas disease, and HIV; toxic causes such as alcohol, chemotherapeutic agents, and peripartum states; metabolic causes including hyperthyroidism, pheochromocytoma, and stress-induced (Takotsubo) cardiomyopathy; and systemic diseases such as lupus, scleroderma, neuromuscular disorders, and amyloidosis.
Pediatric-Specific Etiologies
In children, cardiomyopathy may be idiopathic or genetic, including inborn errors of metabolism, neuromuscular disease, malformation syndromes, and familial isolated cardiomyopathies. Acquired causes include nutritional deficiencies, electrolyte and endocrine disturbances, toxins, collagen vascular disease, immunologic disorders, malignancy, morbid obesity, myocarditis, pulmonary disease, Kawasaki disease, infections, radiation, congenital heart disease, and perinatal asphyxia.
Clinical Manifestations
History may reveal antecedent viral illness, chemotherapy exposure, HIV, Lyme disease, pregnancy, substance use, or systemic conditions such as hemochromatosis or sarcoidosis. Family history of sudden cardiac death is a critical clue. Symptoms include exertional dyspnea, dizziness, palpitations, near-syncope or syncope, ventricular arrhythmias, and congestive heart failure. Pediatric presentations may include irritability, hepatomegaly, generalized weakness, hypoglycemia, metabolic acidosis, hyperammonemia, cyanosis, encephalopathy, and dysmorphic features. Pregnancy-related cardiomyopathy should be considered separately.
Initial Assessment Priorities
Evaluation focuses on vital signs, cardiopulmonary examination, evidence of volume overload, abdominal organomegaly, peripheral edema, and signs of systemic disease such as rash or goiter. Rapid identification of decompensated heart failure, malignant arrhythmias, or shock is essential.
Diagnostic Evaluation
Laboratory studies include CBC, metabolic panel, liver and thyroid function tests, cardiac biomarkers, and BNP (levels >100 pg/mL support heart failure). Serologic testing is rarely helpful in the emergency setting. Chest radiography may demonstrate cardiomegaly, pulmonary congestion, and pleural effusions in dilated cardiomyopathy, whereas restrictive cardiomyopathy often shows a normal cardiac silhouette with pulmonary congestion. Emergency bedside transthoracic echocardiography may reveal depressed LV ejection fraction and exclude pericardial tamponade. Formal echocardiography is the diagnostic study of choice to characterize chamber size, wall thickness, systolic and diastolic function, and valvular disease. CT and cardiac MRI help differentiate restrictive cardiomyopathy from constrictive pericarditis and provide detailed assessment of myocardial anatomy, fibrosis, infiltration, iron overload, and viability.
Electrocardiographic Patterns
Hypertrophic cardiomyopathy commonly shows LV hypertrophy and deep Q waves in inferolateral leads, particularly in adolescents. Dilated, toxic, Lyme, and Chagas cardiomyopathies may present with atrial fibrillation, heart block, conduction delays, or pseudoinfarct patterns. Stress-induced (Takotsubo) cardiomyopathy may mimic STEMI and often necessitates cardiac catheterization to exclude ischemia.
Important Differentials
Other causes of dyspnea include COPD, asthma, anemia, interstitial lung disease, pulmonary embolism, pericardial tamponade, valvular disease, ischemic heart disease, hypothyroidism, and constrictive pericarditis. Syncope differentials include hypovolemia, hypoglycemia, heat illness, arrhythmia, and cardiac ischemia.
Prehospital Considerations
Patients require monitoring, supplemental oxygen, and cautious use of nitrates, particularly in suspected hypertrophic cardiomyopathy. Decompensated heart failure may benefit from nitrates and noninvasive positive-pressure ventilation.
Emergency Stabilization
Management prioritizes airway, breathing, and circulation with oxygen supplementation and noninvasive ventilation when indicated. Intubation is reserved for respiratory failure or severe encephalopathy.
Definitive Emergency Management
Treatment focuses on standard heart failure and arrhythmia protocols. Anticoagulation is indicated in dilated cardiomyopathy with atrial fibrillation or embolic risk. Atrial fibrillation and ventricular dysrhythmias are managed per ACLS principles. In hypertrophic cardiomyopathy, negative inotropes such as beta-blockers or disopyramide may reduce outflow obstruction. Special pediatric considerations include keeping patients NPO until inborn metabolic errors are excluded, administering dextrose-containing fluids cautiously, and avoiding lactate-containing solutions. Carnitine, antioxidants, and vitamin cofactors may be required in metabolic disorders.
Common Emergency Medications
Therapies may include amiodarone, beta-blockers, calcium channel blockers, diuretics, nitrates, anticoagulation, milrinone, nesiritide, and disease-specific agents such as carnitine, with careful age- and comorbidity-adjusted dosing.
Disposition Planning
Admission is required for new or suspected cardiomyopathy, syncope with concern for arrhythmia or hypertrophic cardiomyopathy, family history of sudden death, cardiogenic shock, or significant decompensation. Discharge may be considered for known cardiomyopathy with mild heart failure responding to treatment, typically after cardiology consultation.
Referral and Follow-Up
Patients with reduced ejection fraction (<35%) may require referral for implantable cardioverter-defibrillators, biventricular pacing, ventricular assist devices, or transplant evaluation. ongoing cardiology follow-up and consideration of genetic testing are essential.< />pan>
Clinical Insights
Bedside echocardiography is a powerful emergency tool in patients with syncope or exertional symptoms. A thorough family history is critical, as inherited cardiomyopathies are a major cause of sudden cardiac death in young patients.

- Published on
Emergency and Acute Medicine – Cardiac Transplantation Complications
Overview
Heart transplant recipients represent a high-risk emergency population because of denervated graft physiology and chronic immunosuppression. They are vulnerable to ischemia, heart failure, infection, and medication toxicity. Approximately 1,900–2,300 cardiac transplants are performed annually in the United States, with survival rates of 85–90% at 1 year and about 75% at 5 years. Most patients receive triple-drug immunosuppressive regimens, commonly including corticosteroids. The highest complication rate occurs within the first 6 weeks post-transplant, though late complications are common.
Special Populations
Older transplant recipients have increased susceptibility to severe infection and acute rejection due to age-related immune changes. Pregnancy after cardiac transplantation is increasingly reported and usually results in live births, though hypertension, preeclampsia, and rejection are common. Physiologic changes of pregnancy do not significantly increase heart failure risk, but vigilance for infection and rejection is essential.
Mechanisms of Complications
Rejection may be hyperacute, acute, or chronic. Hyperacute rejection occurs within minutes due to ABO or major incompatibility and is immediately graft-fatal. Acute rejection, characterized by lymphocytic infiltration and myocyte injury, is most common in the first 6 weeks but can occur at any time. Chronic rejection manifests as fibrosis and graft vascular disease and has no effective therapy. Cardiac allograft vasculopathy represents immune-mediated accelerated coronary disease and is the leading cause of late mortality beyond the first year.
Infectious complications dominate early outcomes. During the first month, bacterial infections such as pneumonia, mediastinitis, wound infections, and UTIs are common. Within the first year, opportunistic infections including CMV, HSV, Legionella, fungi, and Pneumocystis become prominent. Medication toxicities are frequent, particularly nephrotoxicity, neurotoxicity, metabolic derangements, and bone marrow suppression from immunosuppressants. Long-term immunosuppression markedly increases malignancy risk, especially skin cancers, lymphomas, Kaposi sarcoma, and solid tumors.
Clinical Manifestations
Acute rejection presents with nonspecific symptoms due to cardiac denervation, including fatigue, dyspnea, low-grade fever, nausea, and vomiting. Heart failure may manifest as tachypnea, rales, hypoxia, S3 gallop, murmurs, and edema. Allograft vasculopathy often presents insidiously with fatigue, cough, or dyspnea; acute presentations include heart failure, infarction, or sudden death, often without angina. Infection may present with fever, skin lesions, or organ-specific symptoms. CMV infection ranges from mild flu-like illness to pneumonitis, hepatitis, gastroenteritis, and severe leukopenia with high mortality. Pediatric patients have higher risk of post-transplant lymphoproliferative disease and pneumonia.
Essential Emergency Assessment
Evaluation focuses on identifying rejection, graft dysfunction, ischemia, and infection. Initial studies include ECG, cardiac enzymes, chest radiograph, and echocardiography. Suspected rejection requires urgent consultation with the transplant team and often endomyocardial biopsy. In children, standard fever evaluation with chest radiograph and ECG is required, and lumbar puncture should be considered if the patient is on steroids.
Diagnostic Findings
Laboratory abnormalities may include renal dysfunction, electrolyte disturbances, metabolic acidosis, and hyperkalemia related to calcineurin inhibitors. Relative eosinophilia may favor rejection over infection. BNP is often chronically elevated. CMV testing and immunosuppressant trough levels are frequently necessary. ECG typically shows sinus tachycardia (90–110 bpm) in denervated hearts; voltage reduction may be seen. Chest radiography may reveal cardiomegaly, pulmonary edema, or effusions. Echocardiography may show diastolic dysfunction, biventricular dilation, valvular regurgitation, or reduced filling parameters.
Key Differentials
Rejection, infection, ischemia, CMV disease, malignancy, and medication toxicity should always be considered.
Prehospital Considerations
Adenosine should be avoided due to prolonged and unpredictable effects in denervated hearts.
Initial Stabilization
Management follows airway, breathing, and circulation priorities with oxygen, IV access, monitoring, and ventilatory or hemodynamic support as needed. Bradycardia does not respond to atropine; isoproterenol is preferred. ACLS protocols apply, with awareness of altered pharmacologic responses.
Emergency Department Management
Hemodynamically significant rejection requires high-dose IV corticosteroids and possible anti–T-cell antibody therapy in coordination with the transplant team. Ischemia or vasculopathy is treated with antiplatelet therapy, anticoagulation, and possible revascularization, though retransplantation may ultimately be required. Suspected CMV infection warrants empiric IV ganciclovir. HSV infections are treated with acyclovir. Fever without source mandates early infectious disease or transplant consultation. Headache or neurologic symptoms require low threshold for neuroimaging and lumbar puncture. Stress-dose steroids may be required during severe illness or trauma, and NSAIDs should be minimized due to nephrotoxicity risk.
Disposition Planning
Admission is indicated for hemodynamically significant rejection, ischemia, new dysrhythmias, heart failure, hypoxia, syncope, poorly controlled hypertension, suspected CMV infection, inability to tolerate oral medications, or fever in immunosuppressed patients. Discharge for mild rejection or select pediatric fever cases should occur only after direct consultation with the transplant team.
Clinical Insights
Symptoms of rejection and infection overlap and are often subtle. Denervated hearts mask classic ischemic pain. Bradycardia will not respond to atropine. Early consultation with the transplant center is essential for optimal outcomes.
Overview
Heart transplant recipients represent a high-risk emergency population because of denervated graft physiology and chronic immunosuppression. They are vulnerable to ischemia, heart failure, infection, and medication toxicity. Approximately 1,900–2,300 cardiac transplants are performed annually in the United States, with survival rates of 85–90% at 1 year and about 75% at 5 years. Most patients receive triple-drug immunosuppressive regimens, commonly including corticosteroids. The highest complication rate occurs within the first 6 weeks post-transplant, though late complications are common.
Special Populations
Older transplant recipients have increased susceptibility to severe infection and acute rejection due to age-related immune changes. Pregnancy after cardiac transplantation is increasingly reported and usually results in live births, though hypertension, preeclampsia, and rejection are common. Physiologic changes of pregnancy do not significantly increase heart failure risk, but vigilance for infection and rejection is essential.
Mechanisms of Complications
Rejection may be hyperacute, acute, or chronic. Hyperacute rejection occurs within minutes due to ABO or major incompatibility and is immediately graft-fatal. Acute rejection, characterized by lymphocytic infiltration and myocyte injury, is most common in the first 6 weeks but can occur at any time. Chronic rejection manifests as fibrosis and graft vascular disease and has no effective therapy. Cardiac allograft vasculopathy represents immune-mediated accelerated coronary disease and is the leading cause of late mortality beyond the first year.
Infectious complications dominate early outcomes. During the first month, bacterial infections such as pneumonia, mediastinitis, wound infections, and UTIs are common. Within the first year, opportunistic infections including CMV, HSV, Legionella, fungi, and Pneumocystis become prominent. Medication toxicities are frequent, particularly nephrotoxicity, neurotoxicity, metabolic derangements, and bone marrow suppression from immunosuppressants. Long-term immunosuppression markedly increases malignancy risk, especially skin cancers, lymphomas, Kaposi sarcoma, and solid tumors.
Clinical Manifestations
Acute rejection presents with nonspecific symptoms due to cardiac denervation, including fatigue, dyspnea, low-grade fever, nausea, and vomiting. Heart failure may manifest as tachypnea, rales, hypoxia, S3 gallop, murmurs, and edema. Allograft vasculopathy often presents insidiously with fatigue, cough, or dyspnea; acute presentations include heart failure, infarction, or sudden death, often without angina. Infection may present with fever, skin lesions, or organ-specific symptoms. CMV infection ranges from mild flu-like illness to pneumonitis, hepatitis, gastroenteritis, and severe leukopenia with high mortality. Pediatric patients have higher risk of post-transplant lymphoproliferative disease and pneumonia.
Essential Emergency Assessment
Evaluation focuses on identifying rejection, graft dysfunction, ischemia, and infection. Initial studies include ECG, cardiac enzymes, chest radiograph, and echocardiography. Suspected rejection requires urgent consultation with the transplant team and often endomyocardial biopsy. In children, standard fever evaluation with chest radiograph and ECG is required, and lumbar puncture should be considered if the patient is on steroids.
Diagnostic Findings
Laboratory abnormalities may include renal dysfunction, electrolyte disturbances, metabolic acidosis, and hyperkalemia related to calcineurin inhibitors. Relative eosinophilia may favor rejection over infection. BNP is often chronically elevated. CMV testing and immunosuppressant trough levels are frequently necessary. ECG typically shows sinus tachycardia (90–110 bpm) in denervated hearts; voltage reduction may be seen. Chest radiography may reveal cardiomegaly, pulmonary edema, or effusions. Echocardiography may show diastolic dysfunction, biventricular dilation, valvular regurgitation, or reduced filling parameters.
Key Differentials
Rejection, infection, ischemia, CMV disease, malignancy, and medication toxicity should always be considered.
Prehospital Considerations
Adenosine should be avoided due to prolonged and unpredictable effects in denervated hearts.
Initial Stabilization
Management follows airway, breathing, and circulation priorities with oxygen, IV access, monitoring, and ventilatory or hemodynamic support as needed. Bradycardia does not respond to atropine; isoproterenol is preferred. ACLS protocols apply, with awareness of altered pharmacologic responses.
Emergency Department Management
Hemodynamically significant rejection requires high-dose IV corticosteroids and possible anti–T-cell antibody therapy in coordination with the transplant team. Ischemia or vasculopathy is treated with antiplatelet therapy, anticoagulation, and possible revascularization, though retransplantation may ultimately be required. Suspected CMV infection warrants empiric IV ganciclovir. HSV infections are treated with acyclovir. Fever without source mandates early infectious disease or transplant consultation. Headache or neurologic symptoms require low threshold for neuroimaging and lumbar puncture. Stress-dose steroids may be required during severe illness or trauma, and NSAIDs should be minimized due to nephrotoxicity risk.
Disposition Planning
Admission is indicated for hemodynamically significant rejection, ischemia, new dysrhythmias, heart failure, hypoxia, syncope, poorly controlled hypertension, suspected CMV infection, inability to tolerate oral medications, or fever in immunosuppressed patients. Discharge for mild rejection or select pediatric fever cases should occur only after direct consultation with the transplant team.
Clinical Insights
Symptoms of rejection and infection overlap and are often subtle. Denervated hearts mask classic ischemic pain. Bradycardia will not respond to atropine. Early consultation with the transplant center is essential for optimal outcomes.

- Published on
Emergency and Acute Medicine – Cardiac Testing
Overview
Cardiac testing is essential in emergency patients with concern for acute coronary syndrome (ACS) or heart failure (HF). These entities exist on a continuum, where unstable angina may progress to myocardial infarction and subsequently to heart failure. Missed ACS remains a major source of emergency department malpractice, with a small but significant percentage of ACS patients discharged inappropriately. History, physical examination, and ECG are the foundation of evaluation but fail to detect a minority of myocardial infarctions, necessitating adjunctive testing such as biomarkers and imaging.
Underlying Causes
ACS most commonly results from atherosclerotic coronary artery disease or coronary vasospasm. In pregnancy, spontaneous coronary artery dissection should be considered when ischemic ECG changes accompany chest pain.
Clinical Presentation
Anginal discomfort is typically provoked by exertion or emotional stress and relieved by rest. Chest pain described as sharp, stabbing, pleuritic, or reproducible by palpation is less suggestive of ACS, though ischemia can still be present in a minority of these cases. Symptom relief with nitroglycerin or gastrointestinal treatments does not reliably distinguish cardiac from noncardiac causes. Anginal episodes often last more than 5 minutes but less than 20 minutes; symptoms persisting beyond 20 minutes raise concern for unstable angina or infarction. Physical examination is frequently normal.
Core Emergency Evaluation
A 12-lead ECG should be obtained within 10 minutes of arrival for patients with chest pain. A single ECG may miss up to half of acute myocardial infarctions, as changes can evolve over time. Hyperacute T waves may represent the earliest ischemic sign. Continuous ECG monitoring or repeat ECGs within 15–60 minutes improves detection. ST-segment depression of 1 mm suggests ischemia, while ST elevation greater than 1–2 mm in contiguous leads defines STEMI. New left bundle branch block complicates diagnosis; concordant ST changes using Sgarbossa criteria increase suspicion. Additional ECG leads may identify infarctions not seen on standard views, including right ventricular and posterior wall involvement.
Laboratory Assessment
Cardiac biomarkers are indicated when ACS is suspected. Troponin I and T rise within 2–3 hours, peak by 8–12 hours, and remain elevated for days. A single negative troponin has limited sensitivity unless drawn 8–12 hours after symptom onset. Mild elevations may occur in nonischemic conditions such as renal failure, sepsis, pulmonary embolism, or heart failure. CK-MB is less sensitive and generally adds little unless renal failure or recent infarction limits troponin interpretation. BNP assists in diagnosing heart failure, with levels above 100 pg/mL supporting the diagnosis.
Imaging Modalities
Chest radiography is often normal but may reveal cardiomegaly, pulmonary edema, pneumonia, or mediastinal widening. Rest echocardiography can identify wall motion abnormalities and pump failure, providing moderate sensitivity for ACS and higher sensitivity for myocardial infarction. Myocardial perfusion imaging using technetium-99m sestamibi detects regions of reduced perfusion and is reserved for intermediate- to high-risk patients. CT coronary angiography offers a high negative predictive value but may increase downstream testing without reducing cost. Exercise stress testing provides diagnostic and prognostic information, with enhanced accuracy when combined with echocardiography or nuclear imaging. Cardiac catheterization remains the gold standard for coronary assessment but does not exclude vasospasm-related events.
Prehospital and Initial ED Management
Prehospital ECG monitoring improves early detection of ACS. In the emergency department, continuous cardiac monitoring and pulse oximetry are standard. Further management depends on risk stratification and test results.
Diagnostic Strategy in the ED
Patients with concerning history should undergo ECG and initial troponin testing. Abnormal findings warrant admission and cardiology consultation. Persistent symptoms or evolving ECG changes necessitate repeat testing or advanced imaging. Low- to moderate-risk patients may undergo stress testing, either during observation or as an outpatient within 72 hours if reliable follow-up exists. Abnormal ancillary testing requires admission or specialty consultation.
Disposition Planning
Admission is indicated for patients with abnormal ECGs, positive biomarkers, concerning imaging, or unavailable observation resources. Observation units may be used for serial testing when diagnosis is uncertain. Discharge is appropriate for patients with noncardiac histories, normal ECGs, and negative cardiac testing.
Follow-Up Considerations
Abnormal stress tests require close cardiology follow-up. Patients discharged with undifferentiated chest pain should have timely outpatient evaluation if inpatient testing is deferred.
Clinical Insights and Cautions
A normal ECG or troponin does not exclude coronary artery disease. Serial ECGs and additional leads increase diagnostic sensitivity. Most emergency patients with undifferentiated chest pain require some form of additional cardiac testing to safely exclude ACS.
Overview
Cardiac testing is essential in emergency patients with concern for acute coronary syndrome (ACS) or heart failure (HF). These entities exist on a continuum, where unstable angina may progress to myocardial infarction and subsequently to heart failure. Missed ACS remains a major source of emergency department malpractice, with a small but significant percentage of ACS patients discharged inappropriately. History, physical examination, and ECG are the foundation of evaluation but fail to detect a minority of myocardial infarctions, necessitating adjunctive testing such as biomarkers and imaging.
Underlying Causes
ACS most commonly results from atherosclerotic coronary artery disease or coronary vasospasm. In pregnancy, spontaneous coronary artery dissection should be considered when ischemic ECG changes accompany chest pain.
Clinical Presentation
Anginal discomfort is typically provoked by exertion or emotional stress and relieved by rest. Chest pain described as sharp, stabbing, pleuritic, or reproducible by palpation is less suggestive of ACS, though ischemia can still be present in a minority of these cases. Symptom relief with nitroglycerin or gastrointestinal treatments does not reliably distinguish cardiac from noncardiac causes. Anginal episodes often last more than 5 minutes but less than 20 minutes; symptoms persisting beyond 20 minutes raise concern for unstable angina or infarction. Physical examination is frequently normal.
Core Emergency Evaluation
A 12-lead ECG should be obtained within 10 minutes of arrival for patients with chest pain. A single ECG may miss up to half of acute myocardial infarctions, as changes can evolve over time. Hyperacute T waves may represent the earliest ischemic sign. Continuous ECG monitoring or repeat ECGs within 15–60 minutes improves detection. ST-segment depression of 1 mm suggests ischemia, while ST elevation greater than 1–2 mm in contiguous leads defines STEMI. New left bundle branch block complicates diagnosis; concordant ST changes using Sgarbossa criteria increase suspicion. Additional ECG leads may identify infarctions not seen on standard views, including right ventricular and posterior wall involvement.
Laboratory Assessment
Cardiac biomarkers are indicated when ACS is suspected. Troponin I and T rise within 2–3 hours, peak by 8–12 hours, and remain elevated for days. A single negative troponin has limited sensitivity unless drawn 8–12 hours after symptom onset. Mild elevations may occur in nonischemic conditions such as renal failure, sepsis, pulmonary embolism, or heart failure. CK-MB is less sensitive and generally adds little unless renal failure or recent infarction limits troponin interpretation. BNP assists in diagnosing heart failure, with levels above 100 pg/mL supporting the diagnosis.
Imaging Modalities
Chest radiography is often normal but may reveal cardiomegaly, pulmonary edema, pneumonia, or mediastinal widening. Rest echocardiography can identify wall motion abnormalities and pump failure, providing moderate sensitivity for ACS and higher sensitivity for myocardial infarction. Myocardial perfusion imaging using technetium-99m sestamibi detects regions of reduced perfusion and is reserved for intermediate- to high-risk patients. CT coronary angiography offers a high negative predictive value but may increase downstream testing without reducing cost. Exercise stress testing provides diagnostic and prognostic information, with enhanced accuracy when combined with echocardiography or nuclear imaging. Cardiac catheterization remains the gold standard for coronary assessment but does not exclude vasospasm-related events.
Prehospital and Initial ED Management
Prehospital ECG monitoring improves early detection of ACS. In the emergency department, continuous cardiac monitoring and pulse oximetry are standard. Further management depends on risk stratification and test results.
Diagnostic Strategy in the ED
Patients with concerning history should undergo ECG and initial troponin testing. Abnormal findings warrant admission and cardiology consultation. Persistent symptoms or evolving ECG changes necessitate repeat testing or advanced imaging. Low- to moderate-risk patients may undergo stress testing, either during observation or as an outpatient within 72 hours if reliable follow-up exists. Abnormal ancillary testing requires admission or specialty consultation.
Disposition Planning
Admission is indicated for patients with abnormal ECGs, positive biomarkers, concerning imaging, or unavailable observation resources. Observation units may be used for serial testing when diagnosis is uncertain. Discharge is appropriate for patients with noncardiac histories, normal ECGs, and negative cardiac testing.
Follow-Up Considerations
Abnormal stress tests require close cardiology follow-up. Patients discharged with undifferentiated chest pain should have timely outpatient evaluation if inpatient testing is deferred.
Clinical Insights and Cautions
A normal ECG or troponin does not exclude coronary artery disease. Serial ECGs and additional leads increase diagnostic sensitivity. Most emergency patients with undifferentiated chest pain require some form of additional cardiac testing to safely exclude ACS.

- Published on
Emergency and Acute Medicine – Cardiac Pacemakers
Core Concept
A cardiac pacemaker is an electronic device that delivers electrical impulses to stimulate myocardial contraction and maintain an adequate heart rate and cardiac output when intrinsic conduction is inadequate.
Pacing Modalities
Transcutaneous pacing uses external pads placed in anterior–lateral or anterior–posterior positions. Current is increased until electrical and mechanical capture is achieved. This is a temporary emergency measure until definitive therapy is available.
Temporary transvenous pacing involves insertion of a pacing wire via central venous access into the right atrium or ventricle, connected to an external generator. It is typically used as a bridge to permanent pacing or until pacing is no longer required.
Permanent implanted pacemakers consist of a lithium-powered battery (lifespan approximately 7–10 years), a programmable generator, and one or more leads positioned in the right atrium and/or ventricle to sense intrinsic activity and pace as needed.
A pacemaker magnet placed over the generator converts the device to asynchronous pacing. This is useful when pacer spikes are absent on ECG; a reduced magnet rate (≈10% decrease) suggests battery depletion.
Key Terminology
Fixed-rate pacing fires at a preset rate regardless of intrinsic rhythm and is now rarely used.
Demand pacing senses native cardiac activity and delivers impulses only when intrinsic rates fall below a programmed threshold.
Sensing refers to the device’s ability to detect intrinsic cardiac depolarization.
Pacemakers are described using a standardized code; in emergency care, the first three letters are most relevant: the chamber sensed, the chamber paced, and the response to sensing. Common configurations include VVI (single-chamber ventricular pacing) and DDD (dual-chamber pacing).
Common Causes of Pacemaker Complications
Infection of pacemaker components is most often due to Staphylococcus epidermidis or Staphylococcus aureus and may be associated with endocarditis; transesophageal echocardiography is preferred for diagnosis.
Venous thrombosis related to pacing leads is common but usually asymptomatic; pulmonary embolism is rare.
Failure to pace may result from lead fracture or disconnection, oversensing of muscle activity or electrical interference, or gradual battery depletion.
Failure to capture is commonly due to lead dislodgment, including Twiddler’s syndrome, or elevated myocardial pacing thresholds caused by hyperkalemia or ischemia.
Pacemaker-mediated tachycardia occurs in dual-chamber devices via re-entry involving the generator and intrinsic conduction system, typically limited to about 140 bpm.
Runaway pacemaker is rare and characterized by extremely rapid pacing (>200 bpm) due to component failure or battery depletion, often causing hemodynamic instability.
Clinical Presentation
Pacemaker failure may manifest as bradycardia, syncope, hypotension progressing to shock, fatigue, dyspnea from heart failure, ischemic chest pain, or altered mental status.
Pacemaker-induced tachycardia presents with dyspnea, chest pain, lightheadedness, or syncope.
Pacemaker syndrome, usually seen with ventricular-only pacing, results from atrioventricular dyssynchrony and causes palpitations, weakness, exercise intolerance, dyspnea, or syncope.
Focused History and Examination
Key history includes date of implantation, device type, and adherence to routine follow-up and battery checks. Examination should include cardiovascular and pulmonary assessment and inspection of the generator site for signs of infection.
Essential Emergency Evaluation
A 12-lead ECG is critical to identify pacing spikes, capture, sensing abnormalities, or dysrhythmias.
Metabolic evaluation should assess for reversible causes of elevated pacing thresholds.
ECG evaluation with a pacemaker magnet helps assess device function: normal magnet rate suggests intact function, a rate slowed by more than 10% indicates battery depletion, and absence of spikes suggests significant malfunction.
Diagnostic Studies
Laboratory testing includes serum potassium, arterial blood gas, and serum levels of antiarrhythmic drugs when relevant.
Chest radiography evaluates lead integrity and position, identifying fractures, dislodgment, or perforation.
Prehospital Considerations
Obtain and preserve rhythm strips for later analysis.
Initial Stabilization
Administer supplemental oxygen, secure the airway if needed, establish IV access, and treat bradycardia per ACLS protocols. Avoid placing defibrillation pads directly over the pacemaker generator. Initiate transcutaneous pacing in unstable patients.
Definitive Emergency Management
For pacemaker failure with instability, initiate transcutaneous pacing and arrange for temporary transvenous pacing via central venous access, preferably the right internal jugular vein. Confirm capture electrically and mechanically, determine capture threshold, and set output at two to three times this threshold.
Correct reversible causes such as hyperkalemia.
Pacemaker-mediated or runaway tachycardia may require AV nodal blockade, device reprogramming, or, in extreme cases, surgical disconnection of the lead.
Medications
Adenosine 6 mg IV bolus may be used in select tachyarrhythmias, with cardiology guidance.
Disposition Planning
Admission is required for permanent pacemaker malfunction or suspected device-related infection.
Patients may be discharged only if asymptomatic and after formal device interrogation by cardiology.
Follow-Up Strategy
All patients require referral to cardiology or a specialized pacemaker clinic for definitive evaluation and management.
Clinical Pearls and Pitfalls
Always consider pacemaker malfunction in patients presenting with unexplained bradycardia, syncope, or decompensated heart failure. Early use of a pacemaker magnet is a simple and valuable diagnostic tool in the emergency setting.
Core Concept
A cardiac pacemaker is an electronic device that delivers electrical impulses to stimulate myocardial contraction and maintain an adequate heart rate and cardiac output when intrinsic conduction is inadequate.
Pacing Modalities
Transcutaneous pacing uses external pads placed in anterior–lateral or anterior–posterior positions. Current is increased until electrical and mechanical capture is achieved. This is a temporary emergency measure until definitive therapy is available.
Temporary transvenous pacing involves insertion of a pacing wire via central venous access into the right atrium or ventricle, connected to an external generator. It is typically used as a bridge to permanent pacing or until pacing is no longer required.
Permanent implanted pacemakers consist of a lithium-powered battery (lifespan approximately 7–10 years), a programmable generator, and one or more leads positioned in the right atrium and/or ventricle to sense intrinsic activity and pace as needed.
A pacemaker magnet placed over the generator converts the device to asynchronous pacing. This is useful when pacer spikes are absent on ECG; a reduced magnet rate (≈10% decrease) suggests battery depletion.
Key Terminology
Fixed-rate pacing fires at a preset rate regardless of intrinsic rhythm and is now rarely used.
Demand pacing senses native cardiac activity and delivers impulses only when intrinsic rates fall below a programmed threshold.
Sensing refers to the device’s ability to detect intrinsic cardiac depolarization.
Pacemakers are described using a standardized code; in emergency care, the first three letters are most relevant: the chamber sensed, the chamber paced, and the response to sensing. Common configurations include VVI (single-chamber ventricular pacing) and DDD (dual-chamber pacing).
Common Causes of Pacemaker Complications
Infection of pacemaker components is most often due to Staphylococcus epidermidis or Staphylococcus aureus and may be associated with endocarditis; transesophageal echocardiography is preferred for diagnosis.
Venous thrombosis related to pacing leads is common but usually asymptomatic; pulmonary embolism is rare.
Failure to pace may result from lead fracture or disconnection, oversensing of muscle activity or electrical interference, or gradual battery depletion.
Failure to capture is commonly due to lead dislodgment, including Twiddler’s syndrome, or elevated myocardial pacing thresholds caused by hyperkalemia or ischemia.
Pacemaker-mediated tachycardia occurs in dual-chamber devices via re-entry involving the generator and intrinsic conduction system, typically limited to about 140 bpm.
Runaway pacemaker is rare and characterized by extremely rapid pacing (>200 bpm) due to component failure or battery depletion, often causing hemodynamic instability.
Clinical Presentation
Pacemaker failure may manifest as bradycardia, syncope, hypotension progressing to shock, fatigue, dyspnea from heart failure, ischemic chest pain, or altered mental status.
Pacemaker-induced tachycardia presents with dyspnea, chest pain, lightheadedness, or syncope.
Pacemaker syndrome, usually seen with ventricular-only pacing, results from atrioventricular dyssynchrony and causes palpitations, weakness, exercise intolerance, dyspnea, or syncope.
Focused History and Examination
Key history includes date of implantation, device type, and adherence to routine follow-up and battery checks. Examination should include cardiovascular and pulmonary assessment and inspection of the generator site for signs of infection.
Essential Emergency Evaluation
A 12-lead ECG is critical to identify pacing spikes, capture, sensing abnormalities, or dysrhythmias.
Metabolic evaluation should assess for reversible causes of elevated pacing thresholds.
ECG evaluation with a pacemaker magnet helps assess device function: normal magnet rate suggests intact function, a rate slowed by more than 10% indicates battery depletion, and absence of spikes suggests significant malfunction.
Diagnostic Studies
Laboratory testing includes serum potassium, arterial blood gas, and serum levels of antiarrhythmic drugs when relevant.
Chest radiography evaluates lead integrity and position, identifying fractures, dislodgment, or perforation.
Prehospital Considerations
Obtain and preserve rhythm strips for later analysis.
Initial Stabilization
Administer supplemental oxygen, secure the airway if needed, establish IV access, and treat bradycardia per ACLS protocols. Avoid placing defibrillation pads directly over the pacemaker generator. Initiate transcutaneous pacing in unstable patients.
Definitive Emergency Management
For pacemaker failure with instability, initiate transcutaneous pacing and arrange for temporary transvenous pacing via central venous access, preferably the right internal jugular vein. Confirm capture electrically and mechanically, determine capture threshold, and set output at two to three times this threshold.
Correct reversible causes such as hyperkalemia.
Pacemaker-mediated or runaway tachycardia may require AV nodal blockade, device reprogramming, or, in extreme cases, surgical disconnection of the lead.
Medications
Adenosine 6 mg IV bolus may be used in select tachyarrhythmias, with cardiology guidance.
Disposition Planning
Admission is required for permanent pacemaker malfunction or suspected device-related infection.
Patients may be discharged only if asymptomatic and after formal device interrogation by cardiology.
Follow-Up Strategy
All patients require referral to cardiology or a specialized pacemaker clinic for definitive evaluation and management.
Clinical Pearls and Pitfalls
Always consider pacemaker malfunction in patients presenting with unexplained bradycardia, syncope, or decompensated heart failure. Early use of a pacemaker magnet is a simple and valuable diagnostic tool in the emergency setting.

- Published on
Emergency And Acute Medicine – Cirrhosis
Overview And Pathophysiology
Cirrhosis is a progressive and chronic disease marked by ongoing inflammation, hepatocellular injury and necrosis, diffuse fibrosis, and the formation of regenerative nodules. These changes lead to distortion and eventual loss of normal hepatic lobular and vascular architecture. In advanced stages, cirrhosis is irreversible and results in intrahepatic portal hypertension due to increased sinusoidal resistance, compression of central veins, and abnormal arterial–portal venous shunting. Cirrhosis is a major public health concern and is among the leading causes of death in the United States.
Etiology And Risk Factors
Chronic alcohol abuse is the most common cause of cirrhosis in the United States, followed by chronic viral hepatitis B and C. Other causes include autoimmune hepatitis, primary biliary cirrhosis, secondary biliary cirrhosis from sclerosing cholangitis, and metabolic disorders such as hereditary hemochromatosis, Wilson disease, and porphyria. Drug-induced liver injury from agents such as acetaminophen, methotrexate, amiodarone, and methyldopa may contribute. Hepatic congestion from right-sided heart failure, constrictive pericarditis, or Budd–Chiari syndrome can lead to cirrhosis. Infiltrative diseases, nonalcoholic steatohepatitis, diffuse hepatocellular carcinoma, and chronic infections such as schistosomiasis or tertiary syphilis are less common causes. Pediatric causes include congenital biliary disorders, metabolic diseases, cystic fibrosis, and α1-antitrypsin deficiency.
Clinical Presentation
Cirrhosis may remain asymptomatic for years or present insidiously with nonspecific complaints such as fatigue, malaise, anorexia, nausea, weight loss, pruritus, and jaundice. Physical findings may include hepatomegaly, splenomegaly, abdominal discomfort, collateral venous circulation with caput medusae, palmar erythema, spider telangiectasias, parotid enlargement, clubbing, and nail changes. Endocrine manifestations include gynecomastia, testicular atrophy, impotence, amenorrhea, and loss of libido. Advanced disease may present with hypotension, fever, fetor hepaticus, asterixis, renal dysfunction, and signs of portal hypertension.
Complications And Decompensated Disease
The development of complications defines decompensated cirrhosis. These include ascites, spontaneous bacterial peritonitis, hepatic encephalopathy, and variceal hemorrhage. Hepatic encephalopathy may be precipitated by gastrointestinal bleeding, infection, excessive dietary protein, electrolyte abnormalities, sedatives, constipation, or renal failure. Variceal bleeding occurs in approximately one-third of patients and carries a high mortality rate with each episode. Other complications include portal hypertensive gastropathy, hepatorenal syndrome due to reduced renal perfusion, and hepatopulmonary syndrome characterized by intrapulmonary vascular dilation and hypoxemia.
Diagnostic Evaluation
Diagnosis relies on a thorough history and physical examination supported by laboratory and imaging studies. Laboratory findings may include anemia, macrocytosis, leukopenia, thrombocytopenia, hypoalbuminemia, hyperbilirubinemia, prolonged prothrombin time, and elevated globulins. Transaminase levels reflect ongoing injury, with an AST to ALT ratio greater than two suggesting alcoholic liver disease. Additional testing is directed at identifying the underlying cause and complications, including viral hepatitis serologies, autoimmune markers, iron studies, ceruloplasmin, α1-antitrypsin levels, and α-fetoprotein. Ultrasound is used to assess liver architecture, ascites, splenomegaly, and portal vein patency, while CT imaging further evaluates abnormal findings. Endoscopy is indicated for variceal surveillance or upper gastrointestinal bleeding, and paracentesis is essential for new or worsening ascites or suspected spontaneous bacterial peritonitis.
Emergency And Acute Management
Initial management focuses on stabilizing the patient and treating life-threatening complications. Altered mental status should prompt administration of dextrose, thiamine, and naloxone as indicated. Hypotension should be corrected with intravenous fluids to prevent ischemic liver injury. Suspected variceal bleeding requires proton pump inhibitors, octreotide infusion, correction of coagulopathy, and urgent endoscopic intervention. Broad-spectrum antibiotics are indicated for suspected sepsis or spontaneous bacterial peritonitis. Hepatic encephalopathy is treated with lactulose and correction of precipitating factors. Ascites management includes sodium restriction, diuretics, and therapeutic paracentesis when necessary.
Definitive And Specialized Therapy
Treatment of underlying etiologies includes phlebotomy or chelation for hemochromatosis, immunosuppressive therapy for autoimmune hepatitis, antiviral therapy for chronic viral hepatitis when appropriate, ursodeoxycholic acid for primary biliary cirrhosis, and chelation therapy for Wilson disease. Nonselective β-blockers reduce the risk of variceal bleeding. Nutritional support with adequate calories and protein is essential unless limited by encephalopathy. Liver transplantation remains the only curative option for advanced cirrhosis.
Disposition And Follow-Up
Patients with acute decompensation, gastrointestinal bleeding, sepsis, advanced hepatic encephalopathy, hepatorenal syndrome, or hepatopulmonary syndrome require hospital admission, often to an intensive care unit. Patients with compensated cirrhosis and no acute complications may be managed as outpatients with close gastroenterology follow-up. All newly diagnosed patients should be referred for specialist evaluation and long-term management.
Clinical Pearls And Pitfalls
Cirrhosis presents with a wide range of clinical manifestations, and prognosis varies significantly depending on disease severity and complications. Any complication signals transition to decompensated disease and worsens prognosis. Symptoms of spontaneous bacterial peritonitis may be subtle, requiring a low threshold for diagnostic paracentesis. Early recognition and aggressive management of complications are critical to improving outcomes.
Overview And Pathophysiology
Cirrhosis is a progressive and chronic disease marked by ongoing inflammation, hepatocellular injury and necrosis, diffuse fibrosis, and the formation of regenerative nodules. These changes lead to distortion and eventual loss of normal hepatic lobular and vascular architecture. In advanced stages, cirrhosis is irreversible and results in intrahepatic portal hypertension due to increased sinusoidal resistance, compression of central veins, and abnormal arterial–portal venous shunting. Cirrhosis is a major public health concern and is among the leading causes of death in the United States.
Etiology And Risk Factors
Chronic alcohol abuse is the most common cause of cirrhosis in the United States, followed by chronic viral hepatitis B and C. Other causes include autoimmune hepatitis, primary biliary cirrhosis, secondary biliary cirrhosis from sclerosing cholangitis, and metabolic disorders such as hereditary hemochromatosis, Wilson disease, and porphyria. Drug-induced liver injury from agents such as acetaminophen, methotrexate, amiodarone, and methyldopa may contribute. Hepatic congestion from right-sided heart failure, constrictive pericarditis, or Budd–Chiari syndrome can lead to cirrhosis. Infiltrative diseases, nonalcoholic steatohepatitis, diffuse hepatocellular carcinoma, and chronic infections such as schistosomiasis or tertiary syphilis are less common causes. Pediatric causes include congenital biliary disorders, metabolic diseases, cystic fibrosis, and α1-antitrypsin deficiency.
Clinical Presentation
Cirrhosis may remain asymptomatic for years or present insidiously with nonspecific complaints such as fatigue, malaise, anorexia, nausea, weight loss, pruritus, and jaundice. Physical findings may include hepatomegaly, splenomegaly, abdominal discomfort, collateral venous circulation with caput medusae, palmar erythema, spider telangiectasias, parotid enlargement, clubbing, and nail changes. Endocrine manifestations include gynecomastia, testicular atrophy, impotence, amenorrhea, and loss of libido. Advanced disease may present with hypotension, fever, fetor hepaticus, asterixis, renal dysfunction, and signs of portal hypertension.
Complications And Decompensated Disease
The development of complications defines decompensated cirrhosis. These include ascites, spontaneous bacterial peritonitis, hepatic encephalopathy, and variceal hemorrhage. Hepatic encephalopathy may be precipitated by gastrointestinal bleeding, infection, excessive dietary protein, electrolyte abnormalities, sedatives, constipation, or renal failure. Variceal bleeding occurs in approximately one-third of patients and carries a high mortality rate with each episode. Other complications include portal hypertensive gastropathy, hepatorenal syndrome due to reduced renal perfusion, and hepatopulmonary syndrome characterized by intrapulmonary vascular dilation and hypoxemia.
Diagnostic Evaluation
Diagnosis relies on a thorough history and physical examination supported by laboratory and imaging studies. Laboratory findings may include anemia, macrocytosis, leukopenia, thrombocytopenia, hypoalbuminemia, hyperbilirubinemia, prolonged prothrombin time, and elevated globulins. Transaminase levels reflect ongoing injury, with an AST to ALT ratio greater than two suggesting alcoholic liver disease. Additional testing is directed at identifying the underlying cause and complications, including viral hepatitis serologies, autoimmune markers, iron studies, ceruloplasmin, α1-antitrypsin levels, and α-fetoprotein. Ultrasound is used to assess liver architecture, ascites, splenomegaly, and portal vein patency, while CT imaging further evaluates abnormal findings. Endoscopy is indicated for variceal surveillance or upper gastrointestinal bleeding, and paracentesis is essential for new or worsening ascites or suspected spontaneous bacterial peritonitis.
Emergency And Acute Management
Initial management focuses on stabilizing the patient and treating life-threatening complications. Altered mental status should prompt administration of dextrose, thiamine, and naloxone as indicated. Hypotension should be corrected with intravenous fluids to prevent ischemic liver injury. Suspected variceal bleeding requires proton pump inhibitors, octreotide infusion, correction of coagulopathy, and urgent endoscopic intervention. Broad-spectrum antibiotics are indicated for suspected sepsis or spontaneous bacterial peritonitis. Hepatic encephalopathy is treated with lactulose and correction of precipitating factors. Ascites management includes sodium restriction, diuretics, and therapeutic paracentesis when necessary.
Definitive And Specialized Therapy
Treatment of underlying etiologies includes phlebotomy or chelation for hemochromatosis, immunosuppressive therapy for autoimmune hepatitis, antiviral therapy for chronic viral hepatitis when appropriate, ursodeoxycholic acid for primary biliary cirrhosis, and chelation therapy for Wilson disease. Nonselective β-blockers reduce the risk of variceal bleeding. Nutritional support with adequate calories and protein is essential unless limited by encephalopathy. Liver transplantation remains the only curative option for advanced cirrhosis.
Disposition And Follow-Up
Patients with acute decompensation, gastrointestinal bleeding, sepsis, advanced hepatic encephalopathy, hepatorenal syndrome, or hepatopulmonary syndrome require hospital admission, often to an intensive care unit. Patients with compensated cirrhosis and no acute complications may be managed as outpatients with close gastroenterology follow-up. All newly diagnosed patients should be referred for specialist evaluation and long-term management.
Clinical Pearls And Pitfalls
Cirrhosis presents with a wide range of clinical manifestations, and prognosis varies significantly depending on disease severity and complications. Any complication signals transition to decompensated disease and worsens prognosis. Symptoms of spontaneous bacterial peritonitis may be subtle, requiring a low threshold for diagnostic paracentesis. Early recognition and aggressive management of complications are critical to improving outcomes.

