|
Pathology - Acute Rejection of Transplant Acute rejection of transplanted kidney, the most prevalent type of transplant rejection. The patient will present with indicators of rejection anywhere from days to up to 6 months post-transplant, which may include myalgias, arthralgias, fever, chills, or other organ-related symptoms such as oliguria for a renal transplant. However, organ-specific function laboratory tests may be the only indication that rejection is occurring, requiring constant monitoring of transplant recipients. The mechanism of acute rejection is a type IV cell-mediated hypersensitivity and concurrent type II antibody-mediated hypersensitivity reaction. Although MHC antigen reactivity was screened for in HLA-matching, HLA complex minor histocompatibility antigens on the allograft (speciesspecific transplant) trigger CD4 + T cells in the recipient to produce cytokines and generate adhesion molecules. This results in proliferation and activation of CD8 + T cells, which release perforins leading to cell death, as well as stimulation of host macrophages by IFN-γ. Adhesion molecules facilitate lymphocyte movement, creating graft edema and endothelial cell damage. The autoantibody reaction is driven by B cell development into plasma cells, producing anti-HLA antibodies, resulting to vasculitis or intimal thickening of the graft. Acute rejection can be controlled with immunosuppressive treatment and is generally reversible. Decline in organ function, which happens many months to years after transplant is chronic rejection. Its mechanism is not well known but results in ischemia to tissue and may be a result of repeated episodes of acute rejection. 
0 Comments
Pathology - Sjogren's Syndrome Sjögren’s syndrome is an autoimmune illness that mostly affects the salivary glands. The primary oral complaint is xerostomia (dry mouth), which can lead to difficulties swallowing, dental caries, and inability to speak continuously. Patients with ocular involvement will commonly complain of a gritty sensation beneath the eyelids, likely from damage of bulbar or corneal conjunctival epithelium due to decreased lacrimation from xerophthalmia (dry eye). The physical findings in this patient are typical, including parotid enlargement. Other possible symptoms include weariness, low-grade fever, myalgias, arthralgias, and Raynaud’s phenomenon. The pathophysiology is a result of exocrine (e.g., salivary, lacrimal) gland invasion by T- and B-cell lymphocytes and B-lymphocyte hyperactivity. Through a number of biological cascades, T-cells trigger glandular epithelial cell death. Ductal and acinar epithelial cells sustain the continuing immunological reaction by improperly generating proinflammatory cytokines and lymphoattractant chemokines. Patient will often have non–organ-specific autoantibodies including ANA and rheumatoid factor (RF); nevertheless, the presence of anti-Ro/SS-A and anti-La/SS-B autoantibodies is specific for Sjögren’s syndrome. Treatment is mainly supportive; punctal plugs can be used to minimize tear drainage into the nasolacrimal duct, and regular dental care should be established. 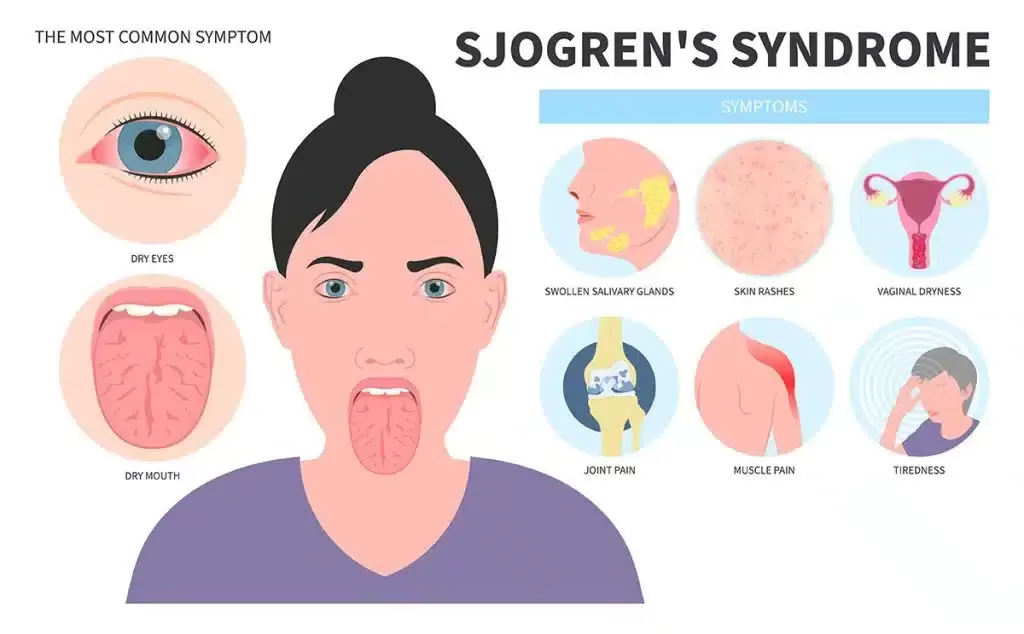 Pathology - Systemic Lupus Erythematosus systemic lupus erythematosus (SLE) is an inflammatory autoimmune illness that affects numerous organ systems. The most common manifestations are skin rashes (as this patient’s characteristic malar butterfly rash), joint pains, and constitutional symptoms; however, renal, cardiac, pulmonary, and neurologic sequelae are also common (mnemonic – SOAP BRAIN MD: serositis, oral ulcers, arthritis, photosensitivity, blood disorders, renal dysfunction, ANA positive, immunologic phenomena, neurologic symptoms, malar rash and discoid rash). It is significantly more common in females and incidence is higher in the African American community. The pathogenesis of SLE likely starts with interaction of susceptibility genes and environmental variables resulting in aberrant immune responses. Immune responses, which may differ between patients, result in the formation of antigens, autoantibodies and immune complexes (type III hypersensitivity response), which circulate and deposit in tissue, resulting in inflammatory processes and damage to multisystem organs. The presence of ANA is frequent in virtually all patients with SLE, but not specific for SLE. However, follow-up testing for anti-dsDNA or anti-Smith antibodies (anti-Sm) is specific for SLE and confirms the diagnosis. The most common cardiac symptom is pericarditis, which responds favorably to NSAIDs. Headaches are very prevalent and periods of psychosis can occasionally occur, but must be separated from glucocorticoidinduced psychosis as immunosuppression is a cornerstone of care of SLE. Diffuse proliferative glomerulonephritis is the most prevalent renal manifestation and the major cause of morbidity and mortality. Anticardiolipin antibodies may also be present and will generate a false-positive in syphilis screening.  Pathology - C5 Deficiency Disorder of the complement system specifically C5 deficiency, leading to bacterial meningitis from Neisseria meningitidis . Complement deficits can result in disruption of the traditional route (C1q, C1r, C1s, C2, and C4), which result in respiratory tract or tissue-invasive bacterial infections, usually by Streptococcus pneumoniae , and can culminate in septicemia. Defects in the alternative pathway (C5, C6, C7, C8, and C9) result in susceptibility to N. gonorrhoeae or N. meningitidis infections. In this example, the patient’s lethargy, fever, vomiting, headache, and positive Brudzinski’s sign (reflexive hip flexion with passive neck flexion) hint toward meningitis. A petechial eruption is symptomatic of N. meningitidis infection and gram-negative diplococci in the CSF corroborate the diagnosis. Functional assays can examine the status of classic and alternative complement pathways, and determination of relative components status can provide a diagnosis. Rarely, a life-threatening and often deadly complication known as Waterhouse–Friderichsen syndrome can emerge due to bleeding within the adrenal glands attributable to N. meningitidis infection.  Pathology - Leukocyte Adhesion Defect Leukocyte adhesion defect is a rare autosomal recessive disorder that comes from mutations of either integrin-mediated leukocyte adhesion (β2 integrin subunit deficiency) or selectin-mediated leukocyte rolling. Thus, neutrophils are not able to connect to the endothelium and undergo diapedesis to reach affected tissues, hence patients are susceptible to bacterial and fungal infections. Significantly high amounts of blood leukocytes are discovered due to the inability of neutrophils to escape the blood arteries. Impaired wound healing with the absence of pus is characteristic. A crucial discovery in this condition is delayed loss of the umbilical chord. Hematopoietic stem cell transplantation may be required in severe types. Currently, bone marrow transplantation is the only curative treatment, however gene therapy may be a possibility in the future.  Pathology - Chronic Granulomatous Disease Chronic granulomatous disease (CGD) is an X-linked recessive condition. Like with other autoimmune disorders, the sorts of microorganisms that infect the patient hint to the deficiency in the immune response. A patient with CGD will have recurrent infections from fungus and staphylococcus (gram-positive cocci in clusters). The deficiency in CGD is inadequate nicotine adenine dinucleotide phosphate oxidase in neutrophil and monocyte cell membranes, resulting to suppressed superoxide dismutase activity and lowered hydrogen peroxide levels. This limits free-radical generation and resulting in an absence respiratory burst, such that phagocytized catalase-positive bacteria are not destroyed. The diagnosis of CGD can be made by the laboratory observation of neutrophils and macrophages failing to decrease a nitroblue tetrazolium dye (leukocytes with an intact respiratory burst create a blue color upon incubation). Myeloperoxidase deficiency, which is a more common abnormality, is characterized by a normal respiratory burst but absence of hypochlorous acid (bleach) synthesis. The only clinical importance, however, shows in vulnerable patients with DM through significant candidal infections.  Pathology - DiGeorge Syndrome congenital thymic aplasia is more often known as DiGeorge syndrome. The condition originates from a failure of the third and fourth pharyngeal pouches to form, resulting in absence of the thymus and parathyroid glands. CATCH-22 is a famous mnemonic used to memorize the clinical signs of the condition. The primary findings are connected to C ardiac defects, as suggested by this patient’s typical ventricular septal defect murmur, as well as A bnormal midline features, including the facial traits mentioned in this patient. Failure of T hymic development results in absence of T cells and susceptibility to opportunistic pathogens, particularly P. jirovecii . C left palate is also a common finding. The lack of PTH can generate symptomatic H ypocalcemia and tetany, as evidenced by the positive Trousseau’s sign (carpal and phalangeal spasm) when the blood pressure cuff is inflated on the patient. The normal genetic mutation is not inherited, but rather a random deletion of the long arm on chromosome 22 (22q11.2 deletion). Live vaccines (e.g., measles, small pox, varicella) are contraindicated. Presentation can be as severe as those of SCID. Therapy involves antimicrobial prophylaxis and thymic transplantation, however the risk of graft-versus-host disease (GVHD) is substantial.  Pathology - Ataxia Telangiectasia Syndrome Ataxia-telangiectasia syndrome is an autosomal recessive disorder stemming from a deficiency in DNA damage repair. Specifically, the ataxia-telangiectasia gene (ATM) is mutated. This gene encodes a protein, which normally serves as a regulator of cell-cycle checkpoints to give time for repair of double-stranded DNA breaks. This abnormality results in deficient cellular immunity and humoral immunodeficiency with thymic hypoplasia due to the fragile chromosomes that are created within the cells. Instability of the chromosomes also puts patients at increased risk for lymphomas and leukemias. Patients will typically present after the first year of life, when they will be delayed in walking secondary to ataxia. The ataxia occurs subsequent to atrophy of the cerebellum, which normally functions to coordinate balance (in conjunction with the vestibular system) and fine motor control. Oculomotor apraxia, which is difficulties with coordinated head and eye movements, can appear as an inability to track objects in young children. Speech will likely also fail to develop adequately. Patients will exhibit characteristic ocular and cutaneous telangiectasias, which are dilated blood vessels. In addition to decreased IgE and IgA, they also interestingly have raised serum alpha-fetoprotein. Patients with Friedrich’s ataxia, which can present similarly, can be separated from ataxia-telangiectasia syndrome because those patients will not have signs of oculomotor apraxia or the immunologic abnormalities.  Pathology - Wiskott - Aldrich Syndrome Symptomatic triad of Wiskott–Aldrich syndrome (WAS) involves T hrombocytopenia, recurrent sinopulmonary I nfections, and E czema (mnemonic: TIE). WAS is an X-linked recessive disorder characterized by poor cell-mediated immunity and decreased IgM production through gradual deletion of T and B cells. The pathogenesis comes from a null-mutation in the gene that codes the WAS protein (WASP), a regulator of actin polymerization in hematopoietic cells. This leads in the absence of a particular glycoprotein receptor on T cells and platelets, resulting to a poor response to polysaccharide vaccines and to thrombocytopenia, respectively. Megakaryocyte levels are typical. Splenomegaly can arise attributable to splenic sequestration of platelets, due to the faulty receptor and so contributing to the thrombocytopenia. Petechiae are small hemorrhages within the skin that are a sign of reduced platelets. Levels of IgG will be normal and IgA and IgE may be normal or elevated. Patients are prone to bacterial, viral, and fungal infections and also have a high frequency of autoimmune diseases and lymphoreticular cancers. Bone marrow transplantation may be useful as treatment. 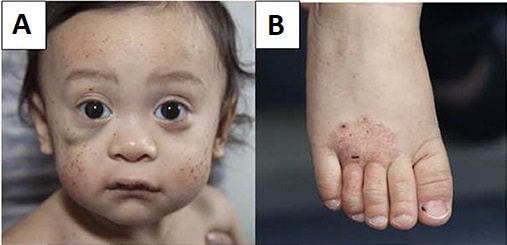 Pathology - Severe Combined Immunodeficiency Disease SCID occurs from a genetic abnormality in stem cells, resulting in absence of the thymus and T and B lymphocytes. In one-half of patients, an autosomal recessive transmission results in the loss of adenosine deaminase, an enzyme involved in purine catabolism. This leads in an accumulation of hazardous deoxyadenosine triphosphate, blocking ribonucleotide reductase and limiting DNA synthesis and therefore lymphocyte proliferation. In other cases, an X-linked transmission leads in absent gamma chain of the interleukin receptor, slowing lymphocyte growth due to issues with interleukin signaling. These patients will present with recurring bacterial, viral, parasite, and fungal infections at a relatively young age; however, maternal antibodies may delay commencement by a few months. Infections will become quite dangerous and include pneumonia, bacteremia, sepsis, and meningitis. Life span is often very short without treatment; however, early discovery and treatment with gene therapy and bone marrow transplantation can be curative.  |
Kembara XtraFacts about medicine and its subtopic such as anatomy, physiology, biochemistry, pharmacology, medicine, pediatrics, psychiatry, obstetrics and gynecology and surgery. Categories
All
|

 RSS Feed
RSS Feed
