|
Pathology - Idiopathic (Immune) Thrombocytopenic Purpura Idiopathic thrombocytopenic purpura (ITP), sometimes called immunological thrombocytopenic purpura, a type II hypersensitivity reaction. This acute onset of the illness most typically occurs in children following a viral upper respiratory tract infection. The pathogenesis relies around the development of IgG antibodies against platelet membrane glycoproteins, notably GpIIb/IIIa receptors. Coating of platelets by IgG leaves them vulnerable to opsonization and phagocytosis by macrophages in the spleen. The lower platelet counts lead to localized cutaneous hemorrhages known as petechiae, described in the vignette, as well as ecchymoses. Other symptoms may include epistaxis, hematuria, or hematochezia. Splenomegaly and lymphadenopathy could be expected in malignant causes of thrombocytopenia. ITP is normally selflimiting in children; nevertheless, severely low levels of platelets (<10,000/mm3) produces risk for serious hemorrhage, bleeding into vital organs, and cerebral hemorrhage. In adults, the problem is most often chronic and is connected with other autoimmune diseases or large array of medications. 
0 Comments
Pathology - Factor V Leiden Thrombophilia Deep venous thrombosis (DVT) in left femoral vein may arise due to factor V Leiden thrombophilia. It is an autosomal dominant inherited disease and the most common hereditary hypercoagulability illness. Family history will likely be positive for venous thromboembolic events (DVT or pulmonary embolism). The condition is a result of mutant clotting factor V that is unable to be destroyed by protein C. Uninhibited factor V activity promotes thrombin production, leading to excess fibrin and excessive coagulation. Patients will likely not have risk factors normally associated with venous thromboembolism, such as prolonged stasis (e.g., recent surgery, long flights, sedentary jobs), hypercoagulability (e.g., malignancy, oral contraceptives, pregnancy), or injury to vasculature (e.g., age, trauma, smoking). The most typical area for clots to form is in veins around the calf; however, advancement to femoral veins is often much more symptomatic. aPC would ordinarily contribute to prolonged coagulation times (aPTT) but will not have this effect in factor V Leiden people. Dilute Russell’s viper venom time may also be used to diagnose factor V Leiden. Clinically, patients with DVT may suffer local soreness to palpation, pain with movement, erythema, warmth, and edema, which may be similar to cellulitis; however, patients with infection will often have a higher fever. Ultrasonography will often pinpoint the thrombus location.  Pathology - Disseminated Intravascular Coagulation (DIC) DIC may occur secondary to gram-negative bacterial septic shock. DIC is a thrombohemorrhagic illness that develops from activation of the coagulation cascade and consumption of coagulation factors as a complication of a number of disorders. Gram-negative organisms (such as N. meningitidis as in this case or most commonly E. coli), trauma, obstetric complications, malignancy or other causes of vascular damage, and inflammatory mediators result in the release or exposure of tissue factor, the most fibrogenic substance known, initiating coagulation. Fibrin thrombi form in the microcirculation, consuming coagulation factors, trapping platelets, and restricting blood flow (producing ischemic tissue damage). Excess thrombin from coagulation results in conversion of plasminogen to plasmin, which cleaves the fibrin thrombi (fibrinolysis) creating fibrin split products and promotes proteolysis of clotting components. The fibrin degradation products (FDPs) in turn exhibit anticoagulant properties, including preventing thrombin production. The net outcome is uncontrollable bleeding. Patients may bleed from any breaks in the skin or mucous membranes. Laboratory results will demonstrate increased PT and PTT, decreased fibrinogen, thrombocytopenia, prolonged bleeding time, increased D-dimer, and presence of FDPs. Fibrin thrombi cause damage to RBC membranes producing a normocytic anemia with schistocytes and reticulocytosis (microangiopathic hemolytic anemia).  Pathology - von Willebrand Disease von Willebrand disease (vWD) is the most common inherited bleeding illness. It is autosomal dominant and is most commonly (in vWD Type I) the outcome of a quantitative deficiency in von Willebrand factor (vWF). vWF is a vital component of hemostasis, forming the adhesive bond between platelets and the damaged endothelium, and also serves as a carrier protein for factor VIII. The condition results in prolonged bleeding time on coagulation testing, as faulty platelet adhesion impedes development of the initial platelet plug, and sometimes prolonged PTT, due to its role in factor VIII availability. Abnormal ristocetin cofactor assay or reduced vWF antigen validates the diagnosis. Patients may initially be recognized via operations such as circumcision, dental work, or surgery. Other manifestations may include easy bruising, excessive bleeding with loss of deciduous teeth or delivery, epistaxis, and menorrhagia. Severe bleeding, such as recurrent hemarthrosis in hemophilia, is unusual. Desmopressin (DDAVP) promotes release of vWF and factor VIII and is a mainstay of therapy, with oral contraceptive pills (estrogen also boosts vWF levels) widely used for menorrhagia.  Pathology - Hemophilia Hemophilia A is a coagulation problem from deficiency in factor VIII, and consequent hemarthrosis of his knee. PTT is prolonged when the intrinsic pathway is damaged; however, in cases of hemarthrosis, PT may also be prolonged as the extrinsic pathway clotting factors are spent in containing a hemorrhage. Hemophilia A is nearly five times more common than hemophilia B (deficiency of factor IX) and is transmitted in an Xlinked recessive manner, however family history may be lacking as 30% of cases are spontaneous mutations. The illnesses are clinically indistinguishable and need testing of serum plasma components. Hemophilia occurs on a range of severity, based on the relative insufficiency of the clotting factor. Less than one percent (<1%) activity is considered serious and results in spontaneous bleeding. Patients with mild hemophilia (5–40% factor activity) bleed heavily at times of trauma and surgery. Those with 1 to less than 5% activity have moderate bleeding episodes. Intracranial hemorrhage is the most common cause of mortality and is commonly spontaneous in moderate and severe illnesses. A typical consequence is joint destruction from recurrent hemarthrosis. In this scenario, an arthrocentesis of the joint would be therapeutic and diagnostic.  Pathology - Hereditary Spherocytosis Hereditary spherocytosis (HS) resulting in the development of calcium bilirubinate (pigment) gallstones and subsequent choledocholithiasis. HS is an autosomal dominant condition resulting in defective proteins (e.g., ankyrin, band 3.1, spectrin) that interact with the RBC skeleton and plasma membrane. Decreased membrane contact leads to small, spherical RBCs with no central pallor (spherocytes). The membrane defect and defective Na+/K+-ATPase pump results in increased permeability to sodium (positive osmotic fragility test as mentioned in the vignette). Extravascular hemolysis of the aberrant cells results in jaundice, splenomegaly, and high indirect (unconjugated) bilirubin levels. Excess bilirubin in bile contributes to production of calcium bilirubinate (black pigment) gallstones. In this example, gallstones have become lodged in the CBD (choledocholithiasis) generating high direct (conjugated) bilirubin and elevated alkaline phosphatase. As the patient is afebrile with negative Murphy’s sign and absent leukocytosis, cholangitis is not anticipated and elective removal of the gallbladder is advised to prevent recurrence and complications. Vaccination for encapsulated organisms is also indicated in patients with autosplenectomy.  Pathology - Sickle-Cell Anemia Sickle-cell anemia is an autosomal recessive disorder most commonly encountered in African Americans. Sickle hemoglobin (HbS) is the result of a missense mutation of valine for glutamic acid at the sixth position of the β-globulin chain. Patients homozygous for the sickle-cell gene (HbSS) do not produce any normal HbA and develop anemia starting around 6 to 9 months of life, resulting from decreased HbF and increased production of HbS. Deoxy-genated hemoglobin molecules cluster and polymerize into needle-like fibers, causing the erythrocytes to assume a sickle shape. Recurrent sickling produces membrane damage and intravascular hemolysis. In addition, aberrant cell shape results in microvascular occlusion, which can trigger pain crises, such as in bones, the lungs, or penis (priapism). Dactylitis (painful swelling of the hands and feet) in infants due to bone infarcts is a common initial presenting symptom; however, the most frequent presentation is painless hematuria. Autosplenectomy (associated with Howell-Jolly bodies present on peripheral smear) often occurs by 2 years of age, which increases risk for infection by encapsulated organisms (e.g., pneumococcus, Neisseria, Salmonella). Family history is frequently notable, like with this child’s uncle who presumably died of sickle-cell problems. The heterozygous condition (HbS, sickle-cell trait) does not result in anemia and conveys resistance to Plasmodium falciparum malaria. 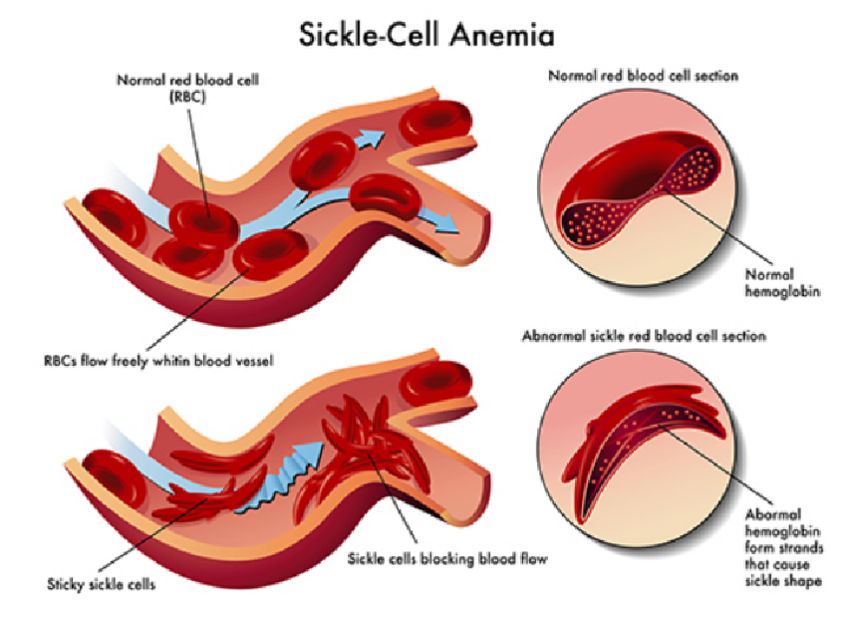 Pathology - Aplastic Anemia Aplastic anemia may occur after carbamazepine medication and subsequently developed invasive aspergillosis. Aplastic anemia originates from defective or inadequate multipotent myeloid stem cells (acquired or hereditary), or antigen receptor change causing T-cell activated stem cell suppression (caused by medications, infection, or other factors). The result is decreased erythrocytes, leukocytes, and platelets, known as pancytopenia. Bone marrow is hypocellular with fatty infiltration. Aplastic anemia is more typically idiopathic, and myelotoxic medicines (alkylating agents, antimetabolites) most commonly employed for cancer therapy generate doserelated suppression. However, certain drugs might result in aplastic anemia in an unanticipated form, including carbamazepine (first-line treatment of TN), chloram-phenicol, streptomycin, hydralazine, and indomethacin to mention a few. Medication side effects might be overwhelming, but it is most important to concentrate on the most prevalent and the most serious. Aplastic anemia can result in significant infection (by opportunistic organisms as in this case) from neutropenia and bleeding from thrombocytopenia. Other causes include certain viruses (parvovirus B19, EBV, HIV) and Fanconi’s anemia.  Pathology - Anemia of Chronic Disease Anemia of chronic disease (ACD) is the most common anemia in hospitalized patients. The patho-genesis can involve decreased EPO production in patients with renal illness; however, more typically, as in this example, chronic inflammatory mediators (e.g., tumor necrosis factor-α, interleukin-1) result in increased synthesis and secretion of hepcidin from the liver. Hepcidin limits the release of iron from macrophages, such that it is sequestered from erythrocyte progenitor cells. As there is no actual deficiency of iron levels in the body, ferritin is still high, and the TIBC is low. However, since iron is being held inside macrophages, serum iron and the transferrin percent saturation is low. Erythrocytes may be either normocytic or microcytic. Inflammatory mediators can also influence EPO, further worsening the anemia. Chronic infections (e.g., TB) can also cause ACD. This patient’s anemia arises from many years of RA and is not related to pneumonia, which she is more susceptible to from her RA medications. The disparity between serum iron and serum ferritin is diagnostic. Although not mentioned in this case, patients with RA are also frequently treated with methotrexate, which inhibits tetrahydrofolate and can lead to folate-deficiency anemia.  Pathology - Folate-Deficiency Anemia Folate-deficiency anemia is the most usually caused by a decreased dietary intake of folate, but can also come from malabsorptive disorders, increased folate usage, and some medicines (5-fluorouracil, methotrexate, trimethoprim-sulfa, phenytoin). Both folate and B12 are involved in the production of deoxythymidine monophosphate. Without production of the nucleic acid thymine, DNA synthesis is impaired, resulting in persistent cell proliferation without division, producing macrocytosis. Thus, RBCs are larger than normal (high MCV) and neutrophils have an increased number of nuclear lobes (hypersegmented neutrophils). Populations at risk for folate-deficiency anemia include those with diets lacking in green vegetables and animal protein, including indigent, elderly (tea and toast diet), and alcoholic patients in particular. Alcohol also hinders the release of folate from the liver, further contributing to the shortfall. Symptoms include those of anemia including fatigue, weakness, headaches, and irritability as well as aphthous ulcers, poor growth, and a swollen tongue. This patient likely has not eaten adequately since the death of his wife. Folate stores normally only last 2 to 3 months. Other indications of self-neglect include absence of appropriate medical care for hypertension and indicators of dehydration (dry mucous membranes and delayed capillary refill).  |
Kembara XtraFacts about medicine and its subtopic such as anatomy, physiology, biochemistry, pharmacology, medicine, pediatrics, psychiatry, obstetrics and gynecology and surgery. Categories
All
|

 RSS Feed
RSS Feed
