|
Pathology - Sickle-Cell Anemia Sickle-cell anemia is an autosomal recessive disorder most commonly encountered in African Americans. Sickle hemoglobin (HbS) is the result of a missense mutation of valine for glutamic acid at the sixth position of the β-globulin chain. Patients homozygous for the sickle-cell gene (HbSS) do not produce any normal HbA and develop anemia starting around 6 to 9 months of life, resulting from decreased HbF and increased production of HbS. Deoxy-genated hemoglobin molecules cluster and polymerize into needle-like fibers, causing the erythrocytes to assume a sickle shape. Recurrent sickling produces membrane damage and intravascular hemolysis. In addition, aberrant cell shape results in microvascular occlusion, which can trigger pain crises, such as in bones, the lungs, or penis (priapism). Dactylitis (painful swelling of the hands and feet) in infants due to bone infarcts is a common initial presenting symptom; however, the most frequent presentation is painless hematuria. Autosplenectomy (associated with Howell-Jolly bodies present on peripheral smear) often occurs by 2 years of age, which increases risk for infection by encapsulated organisms (e.g., pneumococcus, Neisseria, Salmonella). Family history is frequently notable, like with this child’s uncle who presumably died of sickle-cell problems. The heterozygous condition (HbS, sickle-cell trait) does not result in anemia and conveys resistance to Plasmodium falciparum malaria. 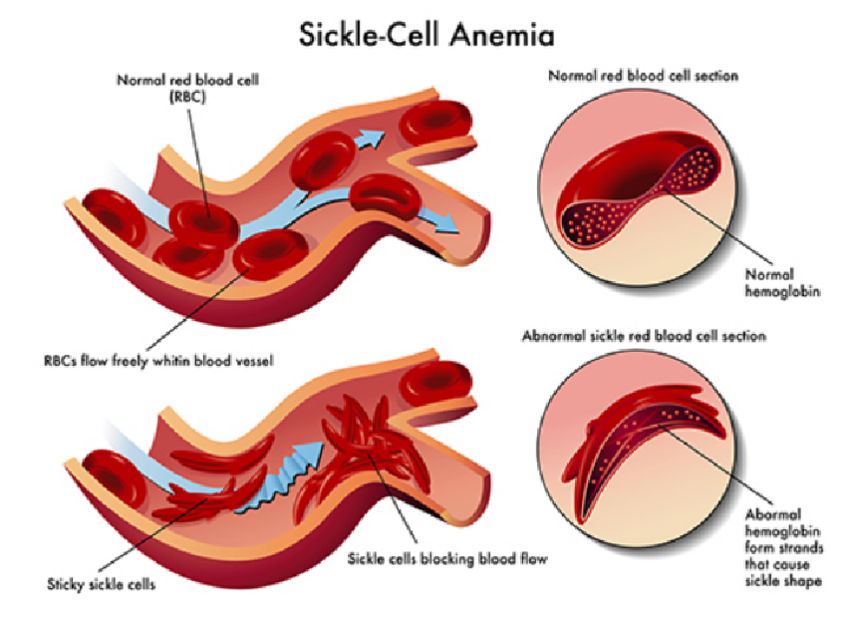
0 Comments
Leave a Reply. |
Kembara XtraFacts about medicine and its subtopic such as anatomy, physiology, biochemistry, pharmacology, medicine, pediatrics, psychiatry, obstetrics and gynecology and surgery. Categories
All
|

 RSS Feed
RSS Feed
