- Published on
Kembara Xtra - Medicine - Neurofibromatosis
Neurocutaneous syndromes (phakomatoses) are neurofibromatosis kinds 1 (NF1) and 2 (NF2). Despite having the same name, they are not related.
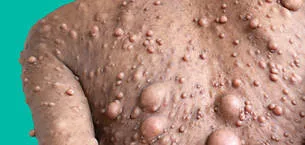
- The neurofibroma, a nerve sheath tumor that grows in close proximity to spinal, peripheral, or cranial nerves, is the distinguishing hallmark of NF1, the most prevalent of the phakomatoses and a multisystem illness that may affect any organ (1). Café au lait macules (CALM), axillary and inguinal freckling, Lisch nodules, and choroidal freckling are further features.
- A uncommon disease called NF2 results in bilateral vestibular schwannomas.
Musculoskeletal, neurological, skin/exocrine, cardiovascular, and neuro-ophthalmologic system(s) affected Synonym(s): von Recklinghausen disease, formerly peripheral NF
Epidemiology
Incidence
Male is more common than female in NF1, and the prevalence is 1:2,500 to 3,000 at birth.
prevalence of one in three to four
Pathophysiology and Etiology
Neurofibromin is a GTPase-activating protein that inhibits p21-ras, a cellular proto-oncogene that promotes cell growth and proliferation, acting as a tumor suppressor.
Biallelic inactivation of NF1 is necessary for several clinical manifestations of NF1, such as CALMs. Others, such malignant peripheral nerve sheath tumors (MPNSTs), are caused by a combination of haploinsufficiency and mutations in other genes, like TP53.
Neurofibromas are benign tumors that form along nerves and are made up of Schwann cells, fibroblasts, mast cells, and vascular components.
Genetics
Autosomal dominant inheritance; brought on by a mutation in the NF1 gene on chromosome 17q11.2; the protein product is known as neurofibromin.
De novo mutations, mostly paternal, are the cause of about 50% of instances; the likelihood rises with paternal age.
If the mutation is known, prenatal diagnostics is an option.
Gene is big (60 exons), and NF1 is caused by more than 3,000 distinct germline mutations. 95% of clinically significant NF1 mutations may be found using molecular technologies, but clinical diagnoses are routinely made in children.
Even within a family, expressivity can vary greatly, although the p.Arg1038Gly missense substitution is associated with a benign phenotype devoid of neurofibromas or other problems.
Risk Elements
A diagnostic requirement for NF1 is having a first-degree relative who is affected, albeit relatives may not be aware that they have NF1.
Those with NF1 who have a positive family history (or a novel mutation) have a 50% chance of passing it on to each child; 1 in 12 children will be badly afflicted.
Accompanying Conditions
Learning difficulties (50–75%), congenital heart disease, pulmonary stenosis, hypertension, and renal artery stenosis
Diagnosis
Clinical characteristics are used to diagnose NF1 in patients. Usually by the age of 4, a diagnosis can be made through routine examination, paying close attention to skin stigmata. A diagnosis of NF1 requires the presence of at least two of the following clinical features: - 6 café au lait (light brown) macules (CALMs), 5 mm in prepubescent individuals or 15 mm in adults - 2 neurofibromas of any type or 1 plexiform (uncircumscribed) neurofibroma - Axillary or inguinal freckling - 2 Lisch nodules (b
History
Having a first-degree relative with NF1 in the family. NF1 symptoms are typically not present at birth, while plexiform neurofibromas and tibial bending are also congenital.
Along with skin lesions, NF1 patients may also experience painful neurofibromas, pathological fractures, or headaches brought on by hypertension brought on by pheochromocytomas.
Optic gliomas can manifest as diencephalic syndrome, squinting, loss of vision, or involuntary eye movement.
clinical assessment
Skin CALMs, which often appear as NF1 before the age of three, develop. 97% of NF1 patients have uniformly pigmented, irregularly shaped light-brown macules; many unaffected people also have one to three of these lesions.
- Neurofibromas: pathognomonic buttonhole invagination, can be hard or soft, cutaneous, subcutaneous, or plexiform. Neurofibromas of the skin typically develop in late childhood or adolescence.
- Up to 50% of neurofibromas are plexiform.
Freckling or hypertrichosis may cover plexiform neurofibromas and impact underlying structures or localised hyperplasia. They are typically congenital and may be inconspicuous in infancy.
The majority of people develop slowly, but some do, particularly in early life.
- Check for new or developing lesions. Skin lesions that are developing quickly need to be examined.
- Axillary freckling (Crowe sign) or inguinal freckling (91%) - Ophthalmologic - Lisch nodules in 30% - Essentially unique to NF, Lisch nodules are asymptomatic; significant only for diagnosis - Pallor or atrophy of optic disc, bulging of orbit, and loss of vision may be signs of optic glioma.
- Choroidal erythema
Skeletal abnormalities include scoliosis and vertebral angulation, as well as localized bone hypertrophy, particularly in the face. Limb abnormalities include pseudoarthrosis of the tibia, tibial dysplasia (anterolateral bowing of the tibia), and nonossifying fibromas of the long bones, which are uncommon but can increase the risk of fracture in adolescents and adults.
Pay close attention to any new focal pain or asymmetries during a neurologic evaluation.
Annual BP measurements. Patients with NF1 are more likely to experience hypertension, which may be brought on by pheochromocytoma, renal artery stenosis, or the aortic stenosis.
Monitor the development of children's brains.
50–75% of people have learning impairments.
Multiple Diagnoses
Geriatric considerations include familial café au lait spots (autosomal dominant, no other NF1 features), Legius syndrome, constitutional mismatch repair deficiency syndrome (CMMRD), NF2, Watson syndrome, LEOPARD syndrome, McCune-Albright syndrome, neurocutaneous melanosis, proteus syndrome, lipomatosis, and Jaffe-Campanacci syndrome.
Child Safety Considerations
Children who inherit the NF1 gene from an afflicted parent typically show external stigmata by the age of one year, however these stigmata may be mild.
If there are no stigmata at age 2, NF is unlikely, but the kid should have another examination. Although young children may have many CALMs but no additional stigmata, a diagnosis can typically be made by the age of 4 years utilizing NIH criteria.
After the age of 10, regular ophthalmologic examinations at increasing intervals.
Molecular confirmation might be necessary, particularly in cases of unusual presentation.
In pediatric populations, there may be a connection between NF1 and lower urinary tract problems. Therefore, a full urologic evaluation should be considered for children displaying indications of lower urinary tract dysfunction (urgency, daytime frequency, nocturia, weak or slow stream, incomplete bladder emptying).
Children with NF1 have been found to have cerebral vascular disorders (migraine, aneurysm, stroke, and cognitive impairments). Early in the diagnostic assessment, screening for cerebrovascular abnormalities may have some prognostic benefit.
Laboratory Results
Confirmatory genetic testing is useful for people who are suspected of having NF1 but do not meet diagnostic criteria, as well as for prenatal diagnosis and preimplantation genetic diagnosis (PGD). Molecular genetic testing is frequently not required for diagnosis.
In 95% of patients with a diagnosis, NF1 gene mutations can be found using molecular genetic testing.
If molecular genetic testing is needed, multistep pathogenic variant detection using cDNA and gDNA sequence analysis is advised. Numerous different mutations in the NF1 gene are the cause of NF1.
Initial examinations (lab, imaging)
Sphenoid dysplasia, long bone cortical thinning, ribbon ribs, and angular scoliosis are typical radiographic findings.
Teenagers' knee radiographs being screened is debatable. Bony changes might be seen on a CT scan.
MRI results for the brain, spine, or orbits (86%). It is debatable whether routine head MRIs of asymptomatic people are necessary. Blindness may result from optic gliomas (on MRI, 11–15%). Despite the fact that areas of enhanced T2 signal intensity (also known as unidentified bright objects) are frequently found on brain MRIs, they are not NF1 diagnostic and most likely have no clinical relevance.
Although modifying diagnostic criteria is explored, the NIH Consensus Development Conference does not advise routine neuroimaging as a method of making a diagnosis.
Other/Diagnostic Procedures
Neuropsychological testing shows intelligence is typically normal but may show significant deficits in language, visuospatial skills, and neuromotor skills. Ophthalmologic evaluation includes slit-lamp examination of the irides and visual field testing to evaluate optic gliomas. Annual screening mammography and surveillance of breast lesions are also recommended.
Management
Initial Line
No particular therapeutic agents are used; instead, symptoms are managed as they appear (e.g., anticonvulsants for seizures, ADHD drugs, blood pressure management, vitamin D supplements for osteopenia in people with low vitamin D levels).
Second Line Several physiologically focused treatments that obstruct the growth of tumors have been put through clinical trials, including mTOR inhibitors, imatinib, and selective MEK inhibitors.
Farnesyltransferase inhibitors, multikinase inhibitors, and antifibrotic medicines have all failed in clinical studies.
Selumetinib is currently the subject of a phase II study in both adults and kids with NF1 and symptomatic plexiform neurofibromas.
A number of NF1 clinical trials are enrolling participants.
Problems to Refer
Patients with increasingly severe NF1 symptoms should be referred to a multidisciplinary NF clinic.
Early referral to orthopedics for congenital tibial bending; referral for psychosocial concerns; educational help for kids with learning impairments or ADHD (40%)
Further Therapies
Laser therapy is not advised for CALMs. The Children's Tumor Foundation (CTF) has formed the NF Clinical Trials Consortium and the CTF NF Clinic Network to support clinical trials. Occupational therapy is indicated for children with NF1 who present with fine motor problems.
Surgical Techniques
Itching, stinging, pain, tenderness, bleeding, and aesthetic issues can all be brought on by dermal neurofibromas.
Surgical removal, laser ablation of tiny lesions, electrodessication, emollients, camouflage makeup, and psychological support are all possible forms of management.
Plexiform neurofibromas, which are typically benign, can lead to pain, deformity, neurologic deficiency, trouble breathing and swallowing, severe hemorrhage, and are susceptible to developing into cancer. The procedure is surgical.
Surgical treatment for malignancy (particularly MPNST) or dystrophic scoliosis (although nondystrophic scoliosis is typically managed conservatively, with bracing).
Take Action
Infancy to one year - Growth and development: mild short stature, macrocephaly (increased brain capacity), aqueductal stenosis/obstructive hydrocephalus - Check for focal neurologic signs or asymmetric neurologic exam, according to the NF1 health monitoring 2008 standards.
ACLMS and axillary freckling have minimal clinical importance. Skeletal anomalies, particularly in the spine and legs.
Annual ophthalmological examination - Brain MRI for visual abnormalities, ongoing headaches, seizures, noticeably larger heads, and plexiform neurofibroma of the head - Assess speech and language for hypernasal speech caused by velopharyngeal insufficiency and delayed expressive language development.
- Annual blood pressure monitoring; developmental examination of learning and motor skills; may benefit from speech/language and/or motor therapy, as well as special education.
5 to 13 years - Check for skin tumors that are deforming the skin and seek advice if surgery is desired to enhance look or function.
- Determine whether puberty has come early or late. Examine for an optic glioma or a hypothalamic lesion if sexual precocity is observed. Review how puberty affects NF.
- Check for ADHD and learning difficulties.
- Assess the student's social development, academic placement, and growth.
- Up until the age of 8, monitor the ophthalmologic status annually; do an eye exam every two years.
- Annual BP monitoring.
- If the patient exhibits symptoms of low self-esteem, refer them to a clinical psychologist or child psychiatrist.
- Talk about how neurofibromas grow during pregnancy and adolescence.
Encourage parents to talk to their children about their diagnoses.
If the adolescent is between the ages of 13 and 21, look for unusual pubertal development.
- Skin testing for plexiform neurofibromas, neurological testing for evidence of deep plexiform neurofibromas, surgical consultation for indications of pressure on deep structures, and yearly blood pressure monitoring.
- Ophthalmologic examination every two years till the age of 18 - Genetic counseling or discussion of NF1 in the family.
- Talk about sexuality, birth control, and other reproductive options.
- If pertinent, discuss how pregnancy affects NF1.
During pregnancy, neurofibromas may grow larger and new tumors may form.
- Examine prenatal diagnosis, or seek the advice of a geneticist.
Education of Patients
Genetic counseling as well as patient education on family planning and potential consequences. Support networks are crucial.
Children's Tumor Foundation, Washington University NF Center, and Neuro Foundation PROGNOSIS Uncertain; the majority of individuals have a moderate form of NF1 and have normal lives. Life expectancy may be cut by 8 to 21 years, and fatalities among people under the age of 40 are more common.
Complications
Skin neurofibromas primarily manifest as disfigurement in exposed regions. With puberty or pregnancy, the number typically rises.
Scoliosis occurs in 10–30% of people (most cases are minor); 2% of long bones bow; osteopenia and osteoporosis; and a big head is frequently but infrequently accompanied by hydrocephalus.
High relative risk (RR) for uncommon malignancies includes increased risk for pheochromocytoma, rhabdomyosarcoma, leukemia, and Wilms tumor. MPNST (5-10%) usually occurs in adults, especially in the area of prior radiotherapy for plexiform neurofibroma. CNS tumors (5-15%), optic pathway glioma (most common), most often asymptomatic but typically presenting before age 6 years if symptomatic
GI neurofibromas may result in GI abnormalities. Neuropsychological phenotype: behavioral issues, executive dysfunction, impaired working memory; reading, numeracy, and visual spatial difficulties.
Seizures: 6–7%. The most typical type of seizure seen is focal, tonic-clonic (60.9%).
Disorders of puberty; frequent adult hypertension; childhood hypertension; childhood hypertension; cord compression and outlet obstruction from pelvic neurofibromas; increased chance of stillbirth; increased risk of prenatal complications;
Neurocutaneous syndromes (phakomatoses) are neurofibromatosis kinds 1 (NF1) and 2 (NF2). Despite having the same name, they are not related.
- The neurofibroma, a nerve sheath tumor that grows in close proximity to spinal, peripheral, or cranial nerves, is the distinguishing hallmark of NF1, the most prevalent of the phakomatoses and a multisystem illness that may affect any organ (1). Café au lait macules (CALM), axillary and inguinal freckling, Lisch nodules, and choroidal freckling are further features.
- A uncommon disease called NF2 results in bilateral vestibular schwannomas.
Musculoskeletal, neurological, skin/exocrine, cardiovascular, and neuro-ophthalmologic system(s) affected Synonym(s): von Recklinghausen disease, formerly peripheral NF
Epidemiology
Incidence
Male is more common than female in NF1, and the prevalence is 1:2,500 to 3,000 at birth.
prevalence of one in three to four
Pathophysiology and Etiology
Neurofibromin is a GTPase-activating protein that inhibits p21-ras, a cellular proto-oncogene that promotes cell growth and proliferation, acting as a tumor suppressor.
Biallelic inactivation of NF1 is necessary for several clinical manifestations of NF1, such as CALMs. Others, such malignant peripheral nerve sheath tumors (MPNSTs), are caused by a combination of haploinsufficiency and mutations in other genes, like TP53.
Neurofibromas are benign tumors that form along nerves and are made up of Schwann cells, fibroblasts, mast cells, and vascular components.
Genetics
Autosomal dominant inheritance; brought on by a mutation in the NF1 gene on chromosome 17q11.2; the protein product is known as neurofibromin.
De novo mutations, mostly paternal, are the cause of about 50% of instances; the likelihood rises with paternal age.
If the mutation is known, prenatal diagnostics is an option.
Gene is big (60 exons), and NF1 is caused by more than 3,000 distinct germline mutations. 95% of clinically significant NF1 mutations may be found using molecular technologies, but clinical diagnoses are routinely made in children.
Even within a family, expressivity can vary greatly, although the p.Arg1038Gly missense substitution is associated with a benign phenotype devoid of neurofibromas or other problems.
Risk Elements
A diagnostic requirement for NF1 is having a first-degree relative who is affected, albeit relatives may not be aware that they have NF1.
Those with NF1 who have a positive family history (or a novel mutation) have a 50% chance of passing it on to each child; 1 in 12 children will be badly afflicted.
Accompanying Conditions
Learning difficulties (50–75%), congenital heart disease, pulmonary stenosis, hypertension, and renal artery stenosis
Diagnosis
Clinical characteristics are used to diagnose NF1 in patients. Usually by the age of 4, a diagnosis can be made through routine examination, paying close attention to skin stigmata. A diagnosis of NF1 requires the presence of at least two of the following clinical features: - 6 café au lait (light brown) macules (CALMs), 5 mm in prepubescent individuals or 15 mm in adults - 2 neurofibromas of any type or 1 plexiform (uncircumscribed) neurofibroma - Axillary or inguinal freckling - 2 Lisch nodules (b
History
Having a first-degree relative with NF1 in the family. NF1 symptoms are typically not present at birth, while plexiform neurofibromas and tibial bending are also congenital.
Along with skin lesions, NF1 patients may also experience painful neurofibromas, pathological fractures, or headaches brought on by hypertension brought on by pheochromocytomas.
Optic gliomas can manifest as diencephalic syndrome, squinting, loss of vision, or involuntary eye movement.
clinical assessment
Skin CALMs, which often appear as NF1 before the age of three, develop. 97% of NF1 patients have uniformly pigmented, irregularly shaped light-brown macules; many unaffected people also have one to three of these lesions.
- Neurofibromas: pathognomonic buttonhole invagination, can be hard or soft, cutaneous, subcutaneous, or plexiform. Neurofibromas of the skin typically develop in late childhood or adolescence.
- Up to 50% of neurofibromas are plexiform.
Freckling or hypertrichosis may cover plexiform neurofibromas and impact underlying structures or localised hyperplasia. They are typically congenital and may be inconspicuous in infancy.
The majority of people develop slowly, but some do, particularly in early life.
- Check for new or developing lesions. Skin lesions that are developing quickly need to be examined.
- Axillary freckling (Crowe sign) or inguinal freckling (91%) - Ophthalmologic - Lisch nodules in 30% - Essentially unique to NF, Lisch nodules are asymptomatic; significant only for diagnosis - Pallor or atrophy of optic disc, bulging of orbit, and loss of vision may be signs of optic glioma.
- Choroidal erythema
Skeletal abnormalities include scoliosis and vertebral angulation, as well as localized bone hypertrophy, particularly in the face. Limb abnormalities include pseudoarthrosis of the tibia, tibial dysplasia (anterolateral bowing of the tibia), and nonossifying fibromas of the long bones, which are uncommon but can increase the risk of fracture in adolescents and adults.
Pay close attention to any new focal pain or asymmetries during a neurologic evaluation.
Annual BP measurements. Patients with NF1 are more likely to experience hypertension, which may be brought on by pheochromocytoma, renal artery stenosis, or the aortic stenosis.
Monitor the development of children's brains.
50–75% of people have learning impairments.
Multiple Diagnoses
Geriatric considerations include familial café au lait spots (autosomal dominant, no other NF1 features), Legius syndrome, constitutional mismatch repair deficiency syndrome (CMMRD), NF2, Watson syndrome, LEOPARD syndrome, McCune-Albright syndrome, neurocutaneous melanosis, proteus syndrome, lipomatosis, and Jaffe-Campanacci syndrome.
Child Safety Considerations
Children who inherit the NF1 gene from an afflicted parent typically show external stigmata by the age of one year, however these stigmata may be mild.
If there are no stigmata at age 2, NF is unlikely, but the kid should have another examination. Although young children may have many CALMs but no additional stigmata, a diagnosis can typically be made by the age of 4 years utilizing NIH criteria.
After the age of 10, regular ophthalmologic examinations at increasing intervals.
Molecular confirmation might be necessary, particularly in cases of unusual presentation.
In pediatric populations, there may be a connection between NF1 and lower urinary tract problems. Therefore, a full urologic evaluation should be considered for children displaying indications of lower urinary tract dysfunction (urgency, daytime frequency, nocturia, weak or slow stream, incomplete bladder emptying).
Children with NF1 have been found to have cerebral vascular disorders (migraine, aneurysm, stroke, and cognitive impairments). Early in the diagnostic assessment, screening for cerebrovascular abnormalities may have some prognostic benefit.
Laboratory Results
Confirmatory genetic testing is useful for people who are suspected of having NF1 but do not meet diagnostic criteria, as well as for prenatal diagnosis and preimplantation genetic diagnosis (PGD). Molecular genetic testing is frequently not required for diagnosis.
In 95% of patients with a diagnosis, NF1 gene mutations can be found using molecular genetic testing.
If molecular genetic testing is needed, multistep pathogenic variant detection using cDNA and gDNA sequence analysis is advised. Numerous different mutations in the NF1 gene are the cause of NF1.
Initial examinations (lab, imaging)
Sphenoid dysplasia, long bone cortical thinning, ribbon ribs, and angular scoliosis are typical radiographic findings.
Teenagers' knee radiographs being screened is debatable. Bony changes might be seen on a CT scan.
MRI results for the brain, spine, or orbits (86%). It is debatable whether routine head MRIs of asymptomatic people are necessary. Blindness may result from optic gliomas (on MRI, 11–15%). Despite the fact that areas of enhanced T2 signal intensity (also known as unidentified bright objects) are frequently found on brain MRIs, they are not NF1 diagnostic and most likely have no clinical relevance.
Although modifying diagnostic criteria is explored, the NIH Consensus Development Conference does not advise routine neuroimaging as a method of making a diagnosis.
Other/Diagnostic Procedures
Neuropsychological testing shows intelligence is typically normal but may show significant deficits in language, visuospatial skills, and neuromotor skills. Ophthalmologic evaluation includes slit-lamp examination of the irides and visual field testing to evaluate optic gliomas. Annual screening mammography and surveillance of breast lesions are also recommended.
Management
Initial Line
No particular therapeutic agents are used; instead, symptoms are managed as they appear (e.g., anticonvulsants for seizures, ADHD drugs, blood pressure management, vitamin D supplements for osteopenia in people with low vitamin D levels).
Second Line Several physiologically focused treatments that obstruct the growth of tumors have been put through clinical trials, including mTOR inhibitors, imatinib, and selective MEK inhibitors.
Farnesyltransferase inhibitors, multikinase inhibitors, and antifibrotic medicines have all failed in clinical studies.
Selumetinib is currently the subject of a phase II study in both adults and kids with NF1 and symptomatic plexiform neurofibromas.
A number of NF1 clinical trials are enrolling participants.
Problems to Refer
Patients with increasingly severe NF1 symptoms should be referred to a multidisciplinary NF clinic.
Early referral to orthopedics for congenital tibial bending; referral for psychosocial concerns; educational help for kids with learning impairments or ADHD (40%)
Further Therapies
Laser therapy is not advised for CALMs. The Children's Tumor Foundation (CTF) has formed the NF Clinical Trials Consortium and the CTF NF Clinic Network to support clinical trials. Occupational therapy is indicated for children with NF1 who present with fine motor problems.
Surgical Techniques
Itching, stinging, pain, tenderness, bleeding, and aesthetic issues can all be brought on by dermal neurofibromas.
Surgical removal, laser ablation of tiny lesions, electrodessication, emollients, camouflage makeup, and psychological support are all possible forms of management.
Plexiform neurofibromas, which are typically benign, can lead to pain, deformity, neurologic deficiency, trouble breathing and swallowing, severe hemorrhage, and are susceptible to developing into cancer. The procedure is surgical.
Surgical treatment for malignancy (particularly MPNST) or dystrophic scoliosis (although nondystrophic scoliosis is typically managed conservatively, with bracing).
Take Action
Infancy to one year - Growth and development: mild short stature, macrocephaly (increased brain capacity), aqueductal stenosis/obstructive hydrocephalus - Check for focal neurologic signs or asymmetric neurologic exam, according to the NF1 health monitoring 2008 standards.
ACLMS and axillary freckling have minimal clinical importance. Skeletal anomalies, particularly in the spine and legs.
Annual ophthalmological examination - Brain MRI for visual abnormalities, ongoing headaches, seizures, noticeably larger heads, and plexiform neurofibroma of the head - Assess speech and language for hypernasal speech caused by velopharyngeal insufficiency and delayed expressive language development.
- Annual blood pressure monitoring; developmental examination of learning and motor skills; may benefit from speech/language and/or motor therapy, as well as special education.
5 to 13 years - Check for skin tumors that are deforming the skin and seek advice if surgery is desired to enhance look or function.
- Determine whether puberty has come early or late. Examine for an optic glioma or a hypothalamic lesion if sexual precocity is observed. Review how puberty affects NF.
- Check for ADHD and learning difficulties.
- Assess the student's social development, academic placement, and growth.
- Up until the age of 8, monitor the ophthalmologic status annually; do an eye exam every two years.
- Annual BP monitoring.
- If the patient exhibits symptoms of low self-esteem, refer them to a clinical psychologist or child psychiatrist.
- Talk about how neurofibromas grow during pregnancy and adolescence.
Encourage parents to talk to their children about their diagnoses.
If the adolescent is between the ages of 13 and 21, look for unusual pubertal development.
- Skin testing for plexiform neurofibromas, neurological testing for evidence of deep plexiform neurofibromas, surgical consultation for indications of pressure on deep structures, and yearly blood pressure monitoring.
- Ophthalmologic examination every two years till the age of 18 - Genetic counseling or discussion of NF1 in the family.
- Talk about sexuality, birth control, and other reproductive options.
- If pertinent, discuss how pregnancy affects NF1.
During pregnancy, neurofibromas may grow larger and new tumors may form.
- Examine prenatal diagnosis, or seek the advice of a geneticist.
Education of Patients
Genetic counseling as well as patient education on family planning and potential consequences. Support networks are crucial.
Children's Tumor Foundation, Washington University NF Center, and Neuro Foundation PROGNOSIS Uncertain; the majority of individuals have a moderate form of NF1 and have normal lives. Life expectancy may be cut by 8 to 21 years, and fatalities among people under the age of 40 are more common.
Complications
Skin neurofibromas primarily manifest as disfigurement in exposed regions. With puberty or pregnancy, the number typically rises.
Scoliosis occurs in 10–30% of people (most cases are minor); 2% of long bones bow; osteopenia and osteoporosis; and a big head is frequently but infrequently accompanied by hydrocephalus.
High relative risk (RR) for uncommon malignancies includes increased risk for pheochromocytoma, rhabdomyosarcoma, leukemia, and Wilms tumor. MPNST (5-10%) usually occurs in adults, especially in the area of prior radiotherapy for plexiform neurofibroma. CNS tumors (5-15%), optic pathway glioma (most common), most often asymptomatic but typically presenting before age 6 years if symptomatic
GI neurofibromas may result in GI abnormalities. Neuropsychological phenotype: behavioral issues, executive dysfunction, impaired working memory; reading, numeracy, and visual spatial difficulties.
Seizures: 6–7%. The most typical type of seizure seen is focal, tonic-clonic (60.9%).
Disorders of puberty; frequent adult hypertension; childhood hypertension; childhood hypertension; cord compression and outlet obstruction from pelvic neurofibromas; increased chance of stillbirth; increased risk of prenatal complications;
0 Comments
