- Published on
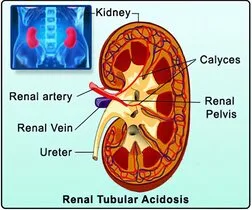
Kembara Xtra - Medicine - Renal Tubular Acidosis
A category of diseases known as renal tubular acidosis (RTA) are defined by the kidney's failure to reabsorb bicarbonate (HCO3) or release hydrogen ions, leading to a normal anion gap metabolic acidosis. There must be normal or almost normal renal function.
Several varieties have been noted:
- Type I (distal) RTA: the distal tubule is unable to acidify the urine due to decreased hydrogen ion secretion, increased hydrogen ion back leak, or decreased sodium reabsorption; the urine pH is greater than 5.5.
- Type II (proximal) RTA: a deficiency in HCO3 reabsorption in the proximal tubule. Urine pH 5.5, no proximal tubular HCO3 reabsorption, compensatory distal HCO3 reabsorption maintains plasma HCO3 levels at 12 to 18 mEq/L.
- Type III RTA: a very rare autosomal recessive condition that is accompanied by intellectual impairment, cerebral calcification, and osteopetrosis.
- Type IV RTA (hypoaldosteronism): caused by a resistance to or a lack in aldosterone, which causes hyperkalemia. The typical pH of urine is 5.5.
Epidemiology
Male is more prevalent than female (in type II RTAs with isolated defects in HCO3 reabsorption).
Pathophysiology and Etiology
Type I RTA is brought on by diseases and drugs that prevent the distal tubule from acidifying urine sufficiently:
Sjögren syndrome, rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and thyroiditis are examples of autoimmune illnesses.Amphotericin B, lithium, ifosfamide, foscarnet, triamterene, trimethoprim, and pentamidine are some examples of medications.
- Hypokalemic obstructive uropathy
Ehlers-Danlos syndrome, glycogenosis type III, Fabry disease, Wilson disease, and other familial disorders
- Hematologic conditions including hereditary elliptocytosis and sickle cell disease (hyperkalemic).
- Toxins: glue and toluene
- Hypergammaglobulinemic syndrome, nephrocalcinosis-causing disorders, hypercalciuria, vitamin D toxicity, and medullary cystic disease
- Primary biliary cirrhosis, chronic active hepatitis, and chronic pyelonephritis
- Incomplete distal RTA, in which individuals are able to excrete enough acid to maintain normal levels of serum HCO3 and pH while being unable to adequately acidify their urine. Voltage-dependent RTA: This type of distal RTA results in retention of K+ and H+ and poor delivery of Na+ to the distal tube, which disrupts the beneficial transepithelial voltage gradient and impairs urine acidification. Amiloride causes voltage-dependent RTA rather than classic distal RTA; this kind will result in hyperkalemia as opposed to hypokalemia in classic distal RTA.
Type II RTA is brought on by illnesses and drugs that prevent the proximal convoluted tubule from reabsorbing HCO3 adequately.
- Genetic pass-through (see below)
Primary Fanconi syndrome, tubulointerstitial nephritis, amyloidosis, multiple myeloma, and other dysproteinemic syndromes are systemic illnesses that can cause Fanconi syndrome. - Medications
: Acetazolamide, methazolamide, and dichlorphenamide are carbonic anhydrase inhibitors. Ifosfamide, oxaliplatin, and cisplatin are chemotherapy drugs. Tenofovir and didanosine are antiretroviral drugs.
Valproic acid and topiramate are anticonvulsants.
Sulfanilamide, out-of-date tetracycline, and aminoglycosides are antibiotics
Additional drugs: deferasirox, apremilast, heavy metals
- Familial (inherited carbonic anhydrase deficiency, tyrosinemia, hereditary fructose intolerance, galactosemia, glycogen storage disease type I, Wilson disease, Lowe syndrome)
- Imbalances in the metabolism of calcium (hyperparathyroidism)
Type IV RTA (5) - NSAIDs, ACE inhibitors, ARBs, low molecular weight (LMW) heparin (hyperkalemia in 5- 10% of patients), ketoconazole, tacrolimus, cyclosporine, spironolactone, eplerenone - Primary adrenal insufficiency - Significantly reduced distal Na+ delivery
- Pseudohypoaldosteronism (PHA), also known as end-organ aldosterone resistance
PHA types 1 and 2 (Gordon syndrome), respectively
Genetics
Intercalated cells in collecting tubules are mutated in Type I RTA, resulting in hereditary variants. Hemolytic anemia, spherocytosis, and ovalocytosis may be brought on by loss-of-function mutations of a chloride-bicarbonate exchanger (AE1) found in the kidney and red blood cell. These mutations are inherited in autosomal dominant and recessive ways. Autosomal recessive loss-of-function mutations of the vacuolar-type H+ ATPase (V-ATPase), which is present in the kidney and inner ear, are linked to larger vestibular aqueducts, vertigo, and sensorineural deafness. More recently, whole exome genomic sequencing has linked the development of distal RTA with early-onset sensorineural deafness to mutations in the protein transcription factor FOXI1, which controls both AE1 and V-ATPase.
Autosomal dominant type II RTAs are incredibly uncommon.
The basolateral electrogenic sodium-bicarbonate cotransporter (NBCe1) mutation is linked to the autosomal recessive type, which is characterized by severe growth retardation, ophthalmologic abnormalities, and intellectual incapacity. A sporadic missense mutation in a sodium phosphate cotransporter can cause Fanconi syndrome, which is linked to a number of genetic diseases (such as cystinosis, Wilson disease, tyrosinemia, hereditary fructose intolerance, Lowe syndrome, galactosemia, glycogen storage disease, and metachromatic leukodystrophy). Other causes of inherited Fanconi syndrome have been discovered in more recent research, including mutations in the genes EHHADH, which is implicated in peroxisomal fatty acid oxidation, and HNF4A, which encodes a nuclear transcription factor.
RTA of Type IV: Some conditions, including PHA type I (autosomal dominant), are hereditary.
Prevention
use and avoidance of harmful substances
Accompanying Conditions
Type I RTA in adults: autoimmune illnesses (Sjögren syndrome, RA, SLE), obstructive uropathy, hypercalciuria Type I RTA in children: hypercalciuria causing rickets, nephrocalcinosis
Type II RTA in adults includes multiple myeloma, carbonic anhydrase inhibitors, aminoglycosides, and Fanconi syndrome (generalized proximal tubular dysfunction resulting in glycosuria, aminoaciduria, hyperuricosuria, phosphaturia, and bicarbonaturia). Type IV RTA includes diabetic nephropathy and solid-organ transplant (due to calcineurin inhibitors).
Diagnoses are frequently asymptomatic (especially type IV).
Rickets and failure to thrive in children
Adult osteomalacia, anorexia, nausea/vomiting, constipation, weakness or polyuria (caused by hypokalemia or hypercalciuria), polydipsia, and
Multiple Diagnoses
Plasma anion gap should be normal; if not, investigate the following conditions: ketoacidosis, ASA, methanol, ethylene glycol, and propylene glycol ingestions; lactic acidosis; D-lactic acidosis; and uremia.
Extrarenal HCO3 losses, chronic diarrhea, fistulas in the small intestine, pancreas, or biliary system, and urinary diversion (such as ureterosigmoidostomy or ileal conduit)
Chronic renal failure-related acidosis (occurs when GFR falls below 20 to 30 mL/min).
Abundant use of chloride salts to administer acid loads, including dilutional acidosis using normal saline (NaCl, HCl, NH4Cl, lysine HCl, CaCl2, MgCl2).
Initial test results from the laboratory and imaging
Electrolytes and serum chemistries
- An typical anion gap in hyperchloremic metabolic acidosis (anion gap = Na+ [Cl + HCO3]).
- Potassium in plasma:
Low: in type I RTA and type II RTA (as a result of decreased distal H+ secretion and increased H+ back leak).
High: for both type I and type IV RTA (if caused by voltagedependent RTA)
- To rule out renal failure as the source of acidosis, BUN and Cr levels should be normal or close to baseline.
- Type II RTA may exhibit electrolyte problems associated with Fanconi syndrome, including hypophosphatemia, hyponatremia, hypoglycemia, and hypoproteinemia.
Studies on urine -
If serum HCO3 is over the resorptive threshold of the distal tubules (12 to 18 mEq/L), urine pH >5.5 in the presence of hyperchloremic metabolic acidosis is strongly suggestive of type I and II RTA. In type IV RTA, urine pH is frequently 5.5 or below.
The presence of urea-splitting organisms and intravascular volume reduction can both affect urine pH, which can mimic RTA nonanion gap metabolic acidosis.
- Type I RTA (3) frequently has excessive urinary calcium levels.
- Urine anion gap (UAG; UNa + UK UCl) is inversely related to urine NH4+ excretion (NH4+ cannot be measured directly in urine). - Increased fractional excretion of phosphate, uric acid, glucose, amino acids, LMW proteins, and presence of urinary retinol-binding protein 4 are sensitive for type II RTA . A positive UAG in an acidemic patient implies poor excretion of urine acid and is indicative of both type I and type IV RTA.
UNa >25 mEq/L is required for accurate UAG. Due to poor acid excretion in renal failure, UAG will also be positive in this condition.
As patients may not excrete the maximum amount of ammonium during acidosis and UAG may not be properly negative, the value of UAG in type II RTA is restricted .
Child Safety Considerations
To assess hearing loss or the existence of dilated vestibular aqueducts in type I RTA, use an audiogram or MRI/CT .
Pregnant women with hereditary type I RTA who presented with severe hypokalemia and metabolic acidosis have been found to have significant acid-base abnormalities. Recurrent urinary tract infections (UTIs), nephrolithiasis brought on by nephrocalcinosis, and problems with alkali supplementation in patients with hyperemesis gravidarum are additional problems that may complicate pregnancy.
Other/Diagnostic Procedures
Use a pH meter on a fresh urine sample for more accurate measurements rather than a dipstick. Adding a coating of oil to the urine will prevent CO2 leakage if the pH cannot be quickly determined.
The presence of non-acidified urine (pH > 5.3) or a positive UAG 6 hours after 100 mg/kg of NH4Cl is diagnostic in the ammonium chloride loading test, which can yield a conclusive diagnosis of type I RTA. Type I RTA is excluded by urine pH 5.3.
- The test's lengthy duration and the gastrointestinal disturbance caused by ammonium chloride's negative effects place restrictions on the study. Patients with type I RTA who are unable to lower their urine pH to 5.3 should consider the concurrent 40 mg furosemide and 1 mg fludrocortisone test (f+f test) as a feasible and safe option.
The gold standard for diagnosing type II RTA is the HCO3 loading test. During a 1 mEq/kg/hr HCO3 infusion, a fractional excretion of HCO3 >15% is diagnostic of type II RTA.
5% does not include type II RTA.
Findings indicate an underlying disease producing RTA, Nephrocalcinosis, Nephrolithiasis, Rickets, Osteomalacia, and Osteopenia
First-line management medication
Administer oral alkali to restore normal serum HCO3 levels. Once HCO3 is normal, increase the dose gradually. Depending on the need for potassium, administer potassium citrate (tablet, powder, or oral solution: 2 mEq K/mL, 2 mEq HCO3/mL), sodium/potassium citrate (oral solution), sodium bicarbonate (NaHCO3) (7.7 mEq NaHCO3/650 mg tab), sodium citrate (1 mEq HCO3 equivalent/mL), or sodium citrate (1 mEq HCO3 equivalent/mL).
Typical doses for Type I RTA are 1 to 2 mEq/kg/day for adults and 3 to 4 mEq/kg/day for children. HCO3 equivalent divided three to four times daily (larger dosages needed if HCO3 waste is prevalent); additional K+ may be needed.Typical doses for Type II RTA are 10 to 15 mEq/kg/day HCO3 equivalent, split 4 to 6 times daily. As renal HCO3 losses rise once plasma HCO3 is corrected over the resorptive threshold, it is very challenging to return plasma HCO3 to normal. Exogenous HCO3 causes an increase in K+ losses, necessitating extra K+; proximal PO4 losses sometimes necessitate supplemental PO4 and vitamin D.
Type IV RTA: Limit dietary K+ and avoid provoking drugs; treat the underlying cause of hypoaldosteronism. You could use polystyrene sulfonate (Kayexalate), thiazide diuretics, or loop diuretics to increase K+ excretion. Correcting hyperkalemia boosts renal ammoniagenesis, the urea cycle, and renal acid excretion by providing more substrate. If necessary, divide 1 to 5 mEq/kg/day of alkali into two to three doses each day. Fludrocortisone: 0.1 to 0.3 mg/day if mineralocorticoid insufficiency.
Safety measures
- Substances containing sodium will increase calcium excretion in the urine, potentially raising the risk of nephrolithiasis.
- Edema and/or hypertension may be brought on by sodium-based alkali and mineralocorticoids.
- If solutions containing citric acid are prescribed, avoid using aluminum-containing drugs (antacids, sucralfate), as citric acid increases the absorption of aluminum.
- Citrate is converted to HCO3 in the liver, whereas NaHCO3 may produce flatulence because CO2 is generated.
Next Line
Thiazide diuretics may be used in type II RTA as supplemental therapy (after maximal alkali replacement) to cause mild hypovolemia. This promotes proximal Na+/HCO3 reabsorption and lowers the amount of necessary alkali replacement, but is likely to further increase urinary K+ losses. They may also be taken into account in type I RTA in order to decrease calcium excretion, but they come with the same danger of hypokalemia.
It is also possible to think about using indomethacin to lessen the severity of polyuria and hypokalemia in type I RTA. Recent case reports and case studies have discussed the security and effectiveness of low-dose fludrocortisone in reducing recurrent hyperkalemia in type IV RTA in the presence of diabetes, amyloidosis, and patients having renal transplant or starting dialysis.
Surgical procedures if obstructive uropathy is the cause of the distal RTA.
Admission
Admit if infant with severe failure to thrive, untrustworthy patient with severe acidosis, or chronic emesis
Patient Follow-Up Monitoring
Poor compliance is typical because to the 3 to 6 times per day alkali dose frequency. Electrolytes should be administered 1 to 2 weeks after the start of therapy, monthly until serum HCO3 has corrected to the appropriate range, and then as clinically required.
Based on the serum K+ level and volume status, the DIET varies.
The prognosis is dependent on the underlying disease:
- Type I RTA: Age-related declines in alkali supplementation are caused by an increased release of H+ from bone throughout skeletal development. Since the primary illness is chronic, alkali replenishment is necessary forever. Children's growth will be restored and nephrocalcinosis will be avoided with therapeutic compliance.
- Type II RTA: Due to the substantial amounts of supplements needed, long-term adherence to therapy is weak.
The prognosis depends on the underlying cause; occasional cases may improve and require no further treatment.
- Treatment and prognosis for Type IV RTA are based on the underlying etiology.
All varieties of RTA may manifest in transitory forms.
Nephrocalcinosis and type I nephrolithiasis,
hypokalemia (type I, type II if given HCO3), type I hypercalciuria, type IV hyperkalemia, and several causes of type I hyperkalemia and Osteopenia (caused by the bone's ability to buffer acid) and osteomalacia (type II caused by phosphate depletion) are complications
A category of diseases known as renal tubular acidosis (RTA) are defined by the kidney's failure to reabsorb bicarbonate (HCO3) or release hydrogen ions, leading to a normal anion gap metabolic acidosis. There must be normal or almost normal renal function.
Several varieties have been noted:
- Type I (distal) RTA: the distal tubule is unable to acidify the urine due to decreased hydrogen ion secretion, increased hydrogen ion back leak, or decreased sodium reabsorption; the urine pH is greater than 5.5.
- Type II (proximal) RTA: a deficiency in HCO3 reabsorption in the proximal tubule. Urine pH 5.5, no proximal tubular HCO3 reabsorption, compensatory distal HCO3 reabsorption maintains plasma HCO3 levels at 12 to 18 mEq/L.
- Type III RTA: a very rare autosomal recessive condition that is accompanied by intellectual impairment, cerebral calcification, and osteopetrosis.
- Type IV RTA (hypoaldosteronism): caused by a resistance to or a lack in aldosterone, which causes hyperkalemia. The typical pH of urine is 5.5.
Epidemiology
Male is more prevalent than female (in type II RTAs with isolated defects in HCO3 reabsorption).
Pathophysiology and Etiology
Type I RTA is brought on by diseases and drugs that prevent the distal tubule from acidifying urine sufficiently:
Sjögren syndrome, rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and thyroiditis are examples of autoimmune illnesses.Amphotericin B, lithium, ifosfamide, foscarnet, triamterene, trimethoprim, and pentamidine are some examples of medications.
- Hypokalemic obstructive uropathy
Ehlers-Danlos syndrome, glycogenosis type III, Fabry disease, Wilson disease, and other familial disorders
- Hematologic conditions including hereditary elliptocytosis and sickle cell disease (hyperkalemic).
- Toxins: glue and toluene
- Hypergammaglobulinemic syndrome, nephrocalcinosis-causing disorders, hypercalciuria, vitamin D toxicity, and medullary cystic disease
- Primary biliary cirrhosis, chronic active hepatitis, and chronic pyelonephritis
- Incomplete distal RTA, in which individuals are able to excrete enough acid to maintain normal levels of serum HCO3 and pH while being unable to adequately acidify their urine. Voltage-dependent RTA: This type of distal RTA results in retention of K+ and H+ and poor delivery of Na+ to the distal tube, which disrupts the beneficial transepithelial voltage gradient and impairs urine acidification. Amiloride causes voltage-dependent RTA rather than classic distal RTA; this kind will result in hyperkalemia as opposed to hypokalemia in classic distal RTA.
Type II RTA is brought on by illnesses and drugs that prevent the proximal convoluted tubule from reabsorbing HCO3 adequately.
- Genetic pass-through (see below)
Primary Fanconi syndrome, tubulointerstitial nephritis, amyloidosis, multiple myeloma, and other dysproteinemic syndromes are systemic illnesses that can cause Fanconi syndrome. - Medications
: Acetazolamide, methazolamide, and dichlorphenamide are carbonic anhydrase inhibitors. Ifosfamide, oxaliplatin, and cisplatin are chemotherapy drugs. Tenofovir and didanosine are antiretroviral drugs.
Valproic acid and topiramate are anticonvulsants.
Sulfanilamide, out-of-date tetracycline, and aminoglycosides are antibiotics
Additional drugs: deferasirox, apremilast, heavy metals
- Familial (inherited carbonic anhydrase deficiency, tyrosinemia, hereditary fructose intolerance, galactosemia, glycogen storage disease type I, Wilson disease, Lowe syndrome)
- Imbalances in the metabolism of calcium (hyperparathyroidism)
Type IV RTA (5) - NSAIDs, ACE inhibitors, ARBs, low molecular weight (LMW) heparin (hyperkalemia in 5- 10% of patients), ketoconazole, tacrolimus, cyclosporine, spironolactone, eplerenone - Primary adrenal insufficiency - Significantly reduced distal Na+ delivery
- Pseudohypoaldosteronism (PHA), also known as end-organ aldosterone resistance
PHA types 1 and 2 (Gordon syndrome), respectively
Genetics
Intercalated cells in collecting tubules are mutated in Type I RTA, resulting in hereditary variants. Hemolytic anemia, spherocytosis, and ovalocytosis may be brought on by loss-of-function mutations of a chloride-bicarbonate exchanger (AE1) found in the kidney and red blood cell. These mutations are inherited in autosomal dominant and recessive ways. Autosomal recessive loss-of-function mutations of the vacuolar-type H+ ATPase (V-ATPase), which is present in the kidney and inner ear, are linked to larger vestibular aqueducts, vertigo, and sensorineural deafness. More recently, whole exome genomic sequencing has linked the development of distal RTA with early-onset sensorineural deafness to mutations in the protein transcription factor FOXI1, which controls both AE1 and V-ATPase.
Autosomal dominant type II RTAs are incredibly uncommon.
The basolateral electrogenic sodium-bicarbonate cotransporter (NBCe1) mutation is linked to the autosomal recessive type, which is characterized by severe growth retardation, ophthalmologic abnormalities, and intellectual incapacity. A sporadic missense mutation in a sodium phosphate cotransporter can cause Fanconi syndrome, which is linked to a number of genetic diseases (such as cystinosis, Wilson disease, tyrosinemia, hereditary fructose intolerance, Lowe syndrome, galactosemia, glycogen storage disease, and metachromatic leukodystrophy). Other causes of inherited Fanconi syndrome have been discovered in more recent research, including mutations in the genes EHHADH, which is implicated in peroxisomal fatty acid oxidation, and HNF4A, which encodes a nuclear transcription factor.
RTA of Type IV: Some conditions, including PHA type I (autosomal dominant), are hereditary.
Prevention
use and avoidance of harmful substances
Accompanying Conditions
Type I RTA in adults: autoimmune illnesses (Sjögren syndrome, RA, SLE), obstructive uropathy, hypercalciuria Type I RTA in children: hypercalciuria causing rickets, nephrocalcinosis
Type II RTA in adults includes multiple myeloma, carbonic anhydrase inhibitors, aminoglycosides, and Fanconi syndrome (generalized proximal tubular dysfunction resulting in glycosuria, aminoaciduria, hyperuricosuria, phosphaturia, and bicarbonaturia). Type IV RTA includes diabetic nephropathy and solid-organ transplant (due to calcineurin inhibitors).
Diagnoses are frequently asymptomatic (especially type IV).
Rickets and failure to thrive in children
Adult osteomalacia, anorexia, nausea/vomiting, constipation, weakness or polyuria (caused by hypokalemia or hypercalciuria), polydipsia, and
Multiple Diagnoses
Plasma anion gap should be normal; if not, investigate the following conditions: ketoacidosis, ASA, methanol, ethylene glycol, and propylene glycol ingestions; lactic acidosis; D-lactic acidosis; and uremia.
Extrarenal HCO3 losses, chronic diarrhea, fistulas in the small intestine, pancreas, or biliary system, and urinary diversion (such as ureterosigmoidostomy or ileal conduit)
Chronic renal failure-related acidosis (occurs when GFR falls below 20 to 30 mL/min).
Abundant use of chloride salts to administer acid loads, including dilutional acidosis using normal saline (NaCl, HCl, NH4Cl, lysine HCl, CaCl2, MgCl2).
Initial test results from the laboratory and imaging
Electrolytes and serum chemistries
- An typical anion gap in hyperchloremic metabolic acidosis (anion gap = Na+ [Cl + HCO3]).
- Potassium in plasma:
Low: in type I RTA and type II RTA (as a result of decreased distal H+ secretion and increased H+ back leak).
High: for both type I and type IV RTA (if caused by voltagedependent RTA)
- To rule out renal failure as the source of acidosis, BUN and Cr levels should be normal or close to baseline.
- Type II RTA may exhibit electrolyte problems associated with Fanconi syndrome, including hypophosphatemia, hyponatremia, hypoglycemia, and hypoproteinemia.
Studies on urine -
If serum HCO3 is over the resorptive threshold of the distal tubules (12 to 18 mEq/L), urine pH >5.5 in the presence of hyperchloremic metabolic acidosis is strongly suggestive of type I and II RTA. In type IV RTA, urine pH is frequently 5.5 or below.
The presence of urea-splitting organisms and intravascular volume reduction can both affect urine pH, which can mimic RTA nonanion gap metabolic acidosis.
- Type I RTA (3) frequently has excessive urinary calcium levels.
- Urine anion gap (UAG; UNa + UK UCl) is inversely related to urine NH4+ excretion (NH4+ cannot be measured directly in urine). - Increased fractional excretion of phosphate, uric acid, glucose, amino acids, LMW proteins, and presence of urinary retinol-binding protein 4 are sensitive for type II RTA . A positive UAG in an acidemic patient implies poor excretion of urine acid and is indicative of both type I and type IV RTA.
UNa >25 mEq/L is required for accurate UAG. Due to poor acid excretion in renal failure, UAG will also be positive in this condition.
As patients may not excrete the maximum amount of ammonium during acidosis and UAG may not be properly negative, the value of UAG in type II RTA is restricted .
Child Safety Considerations
To assess hearing loss or the existence of dilated vestibular aqueducts in type I RTA, use an audiogram or MRI/CT .
Pregnant women with hereditary type I RTA who presented with severe hypokalemia and metabolic acidosis have been found to have significant acid-base abnormalities. Recurrent urinary tract infections (UTIs), nephrolithiasis brought on by nephrocalcinosis, and problems with alkali supplementation in patients with hyperemesis gravidarum are additional problems that may complicate pregnancy.
Other/Diagnostic Procedures
Use a pH meter on a fresh urine sample for more accurate measurements rather than a dipstick. Adding a coating of oil to the urine will prevent CO2 leakage if the pH cannot be quickly determined.
The presence of non-acidified urine (pH > 5.3) or a positive UAG 6 hours after 100 mg/kg of NH4Cl is diagnostic in the ammonium chloride loading test, which can yield a conclusive diagnosis of type I RTA. Type I RTA is excluded by urine pH 5.3.
- The test's lengthy duration and the gastrointestinal disturbance caused by ammonium chloride's negative effects place restrictions on the study. Patients with type I RTA who are unable to lower their urine pH to 5.3 should consider the concurrent 40 mg furosemide and 1 mg fludrocortisone test (f+f test) as a feasible and safe option.
The gold standard for diagnosing type II RTA is the HCO3 loading test. During a 1 mEq/kg/hr HCO3 infusion, a fractional excretion of HCO3 >15% is diagnostic of type II RTA.
5% does not include type II RTA.
Findings indicate an underlying disease producing RTA, Nephrocalcinosis, Nephrolithiasis, Rickets, Osteomalacia, and Osteopenia
First-line management medication
Administer oral alkali to restore normal serum HCO3 levels. Once HCO3 is normal, increase the dose gradually. Depending on the need for potassium, administer potassium citrate (tablet, powder, or oral solution: 2 mEq K/mL, 2 mEq HCO3/mL), sodium/potassium citrate (oral solution), sodium bicarbonate (NaHCO3) (7.7 mEq NaHCO3/650 mg tab), sodium citrate (1 mEq HCO3 equivalent/mL), or sodium citrate (1 mEq HCO3 equivalent/mL).
Typical doses for Type I RTA are 1 to 2 mEq/kg/day for adults and 3 to 4 mEq/kg/day for children. HCO3 equivalent divided three to four times daily (larger dosages needed if HCO3 waste is prevalent); additional K+ may be needed.Typical doses for Type II RTA are 10 to 15 mEq/kg/day HCO3 equivalent, split 4 to 6 times daily. As renal HCO3 losses rise once plasma HCO3 is corrected over the resorptive threshold, it is very challenging to return plasma HCO3 to normal. Exogenous HCO3 causes an increase in K+ losses, necessitating extra K+; proximal PO4 losses sometimes necessitate supplemental PO4 and vitamin D.
Type IV RTA: Limit dietary K+ and avoid provoking drugs; treat the underlying cause of hypoaldosteronism. You could use polystyrene sulfonate (Kayexalate), thiazide diuretics, or loop diuretics to increase K+ excretion. Correcting hyperkalemia boosts renal ammoniagenesis, the urea cycle, and renal acid excretion by providing more substrate. If necessary, divide 1 to 5 mEq/kg/day of alkali into two to three doses each day. Fludrocortisone: 0.1 to 0.3 mg/day if mineralocorticoid insufficiency.
Safety measures
- Substances containing sodium will increase calcium excretion in the urine, potentially raising the risk of nephrolithiasis.
- Edema and/or hypertension may be brought on by sodium-based alkali and mineralocorticoids.
- If solutions containing citric acid are prescribed, avoid using aluminum-containing drugs (antacids, sucralfate), as citric acid increases the absorption of aluminum.
- Citrate is converted to HCO3 in the liver, whereas NaHCO3 may produce flatulence because CO2 is generated.
Next Line
Thiazide diuretics may be used in type II RTA as supplemental therapy (after maximal alkali replacement) to cause mild hypovolemia. This promotes proximal Na+/HCO3 reabsorption and lowers the amount of necessary alkali replacement, but is likely to further increase urinary K+ losses. They may also be taken into account in type I RTA in order to decrease calcium excretion, but they come with the same danger of hypokalemia.
It is also possible to think about using indomethacin to lessen the severity of polyuria and hypokalemia in type I RTA. Recent case reports and case studies have discussed the security and effectiveness of low-dose fludrocortisone in reducing recurrent hyperkalemia in type IV RTA in the presence of diabetes, amyloidosis, and patients having renal transplant or starting dialysis.
Surgical procedures if obstructive uropathy is the cause of the distal RTA.
Admission
Admit if infant with severe failure to thrive, untrustworthy patient with severe acidosis, or chronic emesis
Patient Follow-Up Monitoring
Poor compliance is typical because to the 3 to 6 times per day alkali dose frequency. Electrolytes should be administered 1 to 2 weeks after the start of therapy, monthly until serum HCO3 has corrected to the appropriate range, and then as clinically required.
Based on the serum K+ level and volume status, the DIET varies.
The prognosis is dependent on the underlying disease:
- Type I RTA: Age-related declines in alkali supplementation are caused by an increased release of H+ from bone throughout skeletal development. Since the primary illness is chronic, alkali replenishment is necessary forever. Children's growth will be restored and nephrocalcinosis will be avoided with therapeutic compliance.
- Type II RTA: Due to the substantial amounts of supplements needed, long-term adherence to therapy is weak.
The prognosis depends on the underlying cause; occasional cases may improve and require no further treatment.
- Treatment and prognosis for Type IV RTA are based on the underlying etiology.
All varieties of RTA may manifest in transitory forms.
Nephrocalcinosis and type I nephrolithiasis,
hypokalemia (type I, type II if given HCO3), type I hypercalciuria, type IV hyperkalemia, and several causes of type I hyperkalemia and Osteopenia (caused by the bone's ability to buffer acid) and osteomalacia (type II caused by phosphate depletion) are complications
0 Comments
